




One standard hypothesis of depression is that individuals who are depressed have a lack in monoamine receptors within the body, which in turn leads to reduced levels of neurotransmitters, such as serotonin and norephinephrine, in the brain. But growing evidence supports that at least some kinds of depression might also be linked to continuing low-grade inflammation in the body.
Previous research studies have linked depression with higher level of inflammatory markers when compared with people who aren't depressed. When individuals are given pro-inflammatory cytokines, they report experiencing more symptoms of depression and anxiety. Chronically high levels of inflammation due to health issues, including chronic pain conditions, are also associated with high rates of depression. Even brain imaging of patients with depression show their brain scans have improved neuroinflammation. If your body is in an inflammatory state, fighting off the common cold or influenza, you can experience symptoms overlapping with depression, including, interrupted sleep, depressed mood, fatigue, foggy-headedness and impaired concentration.
A new study published in The Journal of Clinical Psychiatry supports the assumption that an increase in inflammation might play a role in depression. The huge study analyzed data from 14,275 people who have been interviewed between 2007 and 2012 using the Patient Health Questionnaire, or PHQ-9, to screen for depression and had blood samples drawn. They found that people who had depression had 46 percent greater levels of C-reactive protein, or CRP, a marker of inflammatory disease, in their own blood samples. The research study was just able to establish an association between inflammation and depression but not causation, even though it confirms the association of depression with high levels of inflammation as measured through CRP.
The theory that depression might be viewed as a psychoneuroimmunological disorder may also help explain why attempts to reduce chronic inflammation in the body also enhances and helps prevent depression. The purpose of the article below is to demonstrate as well as thoroughly discuss the role of inflammation in depression. Furthermore, the article will describe the evolutionary imperative to the modern treatment target, including a discussion of phytocannabinoids and their association with the treatment of a variety of health issues, including chronic pain symptoms.
The Role of Inflammation in Depression: from Evolutionary Imperative to Modern Treatment Target
Abstract
Introduction
An Evolutionary Perspective
Pathogen Host Defense and Depression
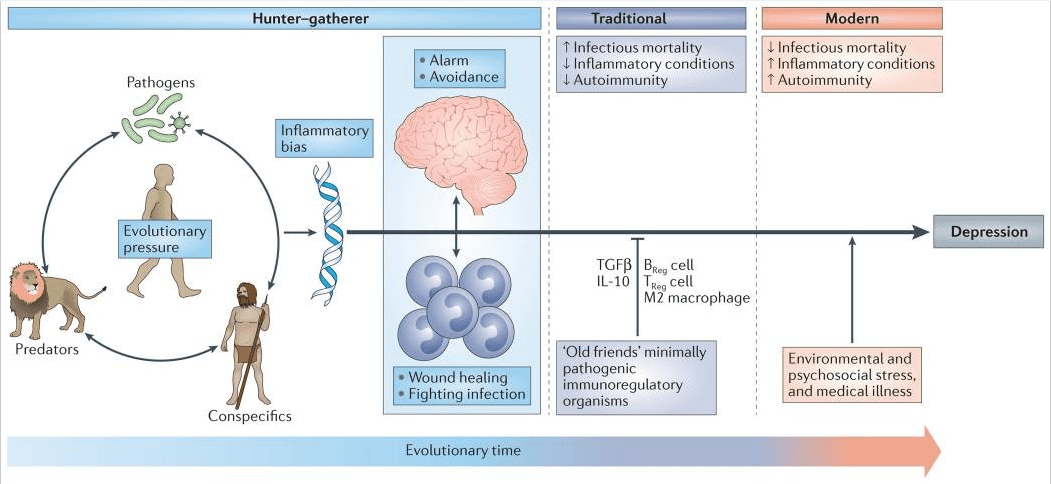
Figure 1: Evolutionary legacy of an inflammatory bias. Early evolutionary pressures derived from human interactions with pathogens, predators and human conspecifics (such as rivals) resulted in an inflammatory bias that included an integrated suite of immunological and behavioural responses that conserved energy for fighting infection and healing wounds, while maintaining vigilance against attack. This inflammatory bias is believed to have been held in check during much of human evolution by exposure to minimally pathogenic, tolerogenic organisms in traditional (that is, rural) environments that engendered immunological responses characterized by the induction of regulatory T (TReg) cells, regulatory B (BReg) cells and immunoregulatory M2 macrophages as well as the production of the anti-inflammatory cytokines interleukin-10 (IL-10) and transforming growth factor-β (TGFβ). In modern times, sanitized urban environments of more developed societies are rife with psychological challenges but generally lacking in the types of infectious challenges that were primary sources of morbidity and mortality across most of human evolution. In the absence of traditional immunological checks and balances, the psychological challenges of the modern world instigate ancestral immunological and behavioural repertoires that represent a decided liability, such as high rates of various inflammation-related disorders including depression.
Most adaptive theories of depression have focused on the potential benefits of depressive symptoms for relationships with other humans7. However, recent models have shifted the focus away from relationships with people, to relationships — both detrimental and beneficial — with pathogens6,8. These theories, which are supported by converging evidence (BOX 1), posit that modern humans have inherited a genomic bias towards inflammation because this response — and the depressive symptoms it promotes — enhanced host survival and reproduction in the highly pathogenic environments in which humans evolved6. From this theoretical perspective, at least some of the human vulnerability to depression evolved out of a behavioural repertoire — often referred to as ‘sickness behaviour’ — which promoted host survival in the face of infection. Indeed, it has been hypothesized that the social avoidance and anhedonia characteristic of depression serve to shunt energy resources to fighting infection and wound healing, whereas the hypervigilance characteristic of anxiety disorders, commonly comorbid with depression, subserves protection from attack and subsequent pathogen exposure6,9. Even psychological stress can be understood from this theoretical perspective, given that the vast majority of stressors faced by mammals over evolutionary time boiled down to risks inherent in hunting, being hunted or competing for reproductive access or status. In all of these circumstances, the risk of pathogen invasion — and subsequent death from infection — was greatly increased as a result of wounding. In ancestral environments, the association between stress perception and risk of subsequent wounding was reliable enough that evolution favoured organisms that prepotently activated inflammatory systems in response to a wide array of environmental threats and challenges (including psychosocial stressors), even if this activation was often a ‘false alarm’ (REF. 6).
Pathogen Host Defense Hypothesis of Depression
- Until recently, approximately 50% of humans died from infectious causes before adulthood, thereby providing strong selective pressure for genetic alleles that enhance host defence124.
- As a result of strong selective pressure, microbial interactions have been a primary driver of human evolution125.
- Patterns of inflammatory activation associated with depression promote survival in highly pathogenic environments while increasing mortality in sanitary conditions common in the developed world126.
- The best replicated risk alleles for depression have pro-inflammatory and/or anti-pathogen protective effects or have been implicated in social behaviours that are likely to reduce pathogen exposure6.
- Environmental risk factors for the development of depression (that is, psychosocial stress, early life adversity, obesity and processed-food diet) are uniformly pro-inflammatory13.
- Exposure to pro-inflammatory cytokines produces a sickness syndrome with symptoms that overlap considerably with those seen in depression and that can be ameliorated by treatment with antidepressants23. In addition, the onset of depression is often mistaken with development of sickness, and symptoms
- associated with infections are often mistaken with the onset of depression127.
- Chronic cytokine exposure produces a combination of withdrawal and/or energy conservation, anxiety and/or hypervigilance behaviours and emotions that commonly coexist in depression6,9.
- Symptoms shared by depression and sickness behaviour — such as hyperthermia and reduced iron availability — that lack any conceivable social value have potent anti-pathogen effects6.
Modern Exaggeration of the Inflammatory Bias
Inflammation and Depression
As the field has matured, it has become increasingly apparent that inflammatory markers are elevated not only in a subgroup of patients with depression30,31 but also in patients with other neuropsychiatric disorders including anxiety disorders and schizophrenia32,33. Moreover, as described below, it may be more accurate to characterize the impact of inflammation on behaviour as being associated not wholly with depression but with specific symptom dimensions across diagnoses that align with the Research Domain Criteria framework put forth by the National Institute of Mental Health (US Department of Health and Human Services). These symptoms, including positive and negative valence systems, relate to altered motivation and motor activity (anhedonia, fatigue and psychomotor impairment) and increased threat sensitivity (anxiety, arousal and alarm)34. Finally, inflammation has been associated with antidepressant treatment non-responsiveness9,32,35–37. For example, in a recent study, 45% of patients with non-response to conventional antidepressants exhibited a CRP >3 mg L−1 (REF. 30), which is considered indicative of a high level of inflammation on the basis of widely accepted cut-off points38. Of note, however, the percentage of patients with high CRP levels can vary as a function of the population being studied, with higher percentages in patients with depression and treatment resistance, childhood maltreatment, medical illnesses and metabolic syndrome.
Immune Pathways Involved in Depression
Inflammasomes: Stress in Translation
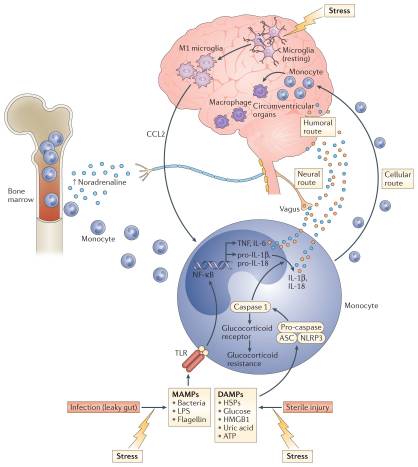
Figure 2: Transmitting stress-induced inflammatory signals to the brain. In the context of psychosocial stress, catecholamines (such as noradrenaline) released by activated sympathetic nervous system fibres stimulate bone marrow production and the release of myeloid cells (for example, monocytes) that enter the periphery where they encounter stress-induced damage-associated molecular patterns (DAMPs), bacteria and bacterial products such as microbial-associated molecular patterns (MAMPs) leaked from the gut. These DAMPs and MAMPs subsequently activate inflammatory signalling pathways such as nuclear factor-κB (NF-κB) and the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome. Stimulation of NLRP3 in turn activates caspase 1, which leads to the production of mature interleukin-1β (IL-1β) and IL-18 while also cleaving the glucocorticoid receptor contributing to glucocorticoid resistance. Activation of NF-κB stimulates the release of other pro-inflammatory cytokines including tumour necrosis factor (TNF) and IL-6, which together with IL-1β and IL-18 can access the brain through humoral and neural routes. Psychosocial stress can also lead to the activation of microglia to a M1 pro-inflammatory phenotype, which release CC-chemokine ligand 2 (CCL2) that in turn attracts activated myeloid cells to the brain via a cellular route. Once in the brain, activated macrophages can perpetuate central inflammatory responses. ASC, apoptosis-associated speck-like protein containing a CARD; HMGB1, high mobility group box 1; HSP, heat shock protein; LPS, lipopolysaccharide; TLR, Toll-like receptor.
Supporting the potential role of the NLRP3 inflammasome in human depression are data demonstrating that increased expression of NLRP3 and caspase 1 in peripheral blood mononuclear cells of patients with depression is associated with increased blood concentrations of IL-1β and IL-18, which in turn correlate with depression severity19,50. In addition, DAMPs that are known to activate NLRP3 are increased in patients with mood disorders, with examples including HSPs, reactive oxygen species and other markers of oxidative stress such as xanthine oxidase, peroxides and F2-isoprostanes51–53. Finally, there is increasing interest in the potential role of the gut microbiome in mood regulation, which may be mediated in part by the inflammasomes54. Indeed, non-pathogenic commensal bacteria and derived microbial-associated molecular patterns (MAMPs) in the gut can leak into the peripheral circulation during stress and activate the inflammasomes55, a process mediated by the SNS and catecholamines56 (FIG. 2). Of note, stress-induced increases in IL-1β and IL-18 were attenuated by treating animals with antibiotics or neutralizing lipopolysaccharide (LPS), demonstrating the importance of the composition of the gut microbiome and gut permeability in stress-induced inflammatory responses55. Taken together, these data support the notion that the inflammasome may be a key immunological point of integration of stress-induced danger signals that ultimately drive inflammatory responses relevant to depression.
Transmitting Inflammatory Signals to the Brain
Work from laboratory animal studies has elucidated several pathways through which inflammatory signals can be transmitted from the periphery to the brain (FIG. 2). These data support the idea that inflammatory responses in peripheral tissues may drive inflammation in the brain leading to depression. Much of the early work focused on how inflammatory cytokines, which are relatively large molecules, could cross the blood–brain barrier (BBB) and influence brain function64. Two major pathways have been described: the ‘humoral pathway’, which involves cytokine passage through leaky regions in the BBB, such as the circumventricular organs, and the binding of cytokines to saturable transport molecules on the BBB; and the ‘neural pathway’, which involves the binding of cytokines to peripheral afferent nerve fibres, such as the vagus nerve, that in turn stimulate ascending catecholaminergic fibres in the brain and/or are translated back into central cytokine signals16. More recently, however, attention has shifted to a third pathway referred to as the ‘cellular pathway’, which involves the trafficking of activated immune cells, typically monocytes, to the brain vasculature and parenchyma. The details of this pathway have been elegantly dissected in the context of behavioural changes in mice that are associated with peripherally induced inflammation in the liver65. In these studies, the release of TNF from inflamed liver was found to stimulate microglial cell production of CC-chemokine ligand 2 (CCL2; also known as MCP1) that then attracted monocytes to the brain65. Blockade of monocyte infiltration to the brain using antibodies specific for the adhesion molecules P-selectin and α4 integrin abrogated depressive-like behaviour in this animal model65. Of note, cytokine-stimulated astrocytes also may be major producers of chemokines, such as CCL2 and CXC-chemokine ligand 1 (CXCL1), that attract immune cells to the brain66. The cellular pathway additionally has been elucidated in the context of social defeat stress, whereby GFP-labelled monocytes coalesced in several regions of the brain associated with the detection of threat (for example, amygdala) — an effect that was dependent on CCL2 and was facilitated by mobilization of monocytes from the bone marrow as a result of stress-induced release of catecholamines67,68 (FIG. 2). Of note, initial microglial activation during social defeat stress appeared to be a result of neuronal activation by catecholamines and decreased neuronal production of CX3C-chemokine ligand 1 (CX3CL1; also known as fractalkine), which maintains microglia in a quiescent state67,68. Interestingly, this cellular pathway has received intriguing support from post-mortem analyses of brain tissue from patients with depression who committed suicide that showed increased numbers of perivascular macrophages in association with increased gene expression of allograft inflammatory factor 1 (AIF1, also known as IBA1) and CCL2, which are associated with macrophage activation and cellular trafficking59.
This evidence of peripheral myeloid cells trafficking to the brain during depression constitutes some of the first data supporting the existence of a central inflammatory response in human depression that is primarily driven by peripheral inflammatory events. Moreover, data demonstrate that antibodies that are specific for TNF but which do not cross the BBB, can block stress-induced depression in mice69. These findings indicate that peripheral inflammatory responses not only can provide important clues to the immunological mechanisms of inflammation in depression but also may serve as biomarkers and targets of immune-based therapies for depression. Protein biomarkers such as plasma CRP and TNF as well as immunotherapies targeting individual cytokines such as TNF, IL-1 and IL-6 may be most relevant in this regard. Of note, plasma CRP is a strong response predictor in anti-cytokine therapy70.
Cytokines and Neurotransmitters
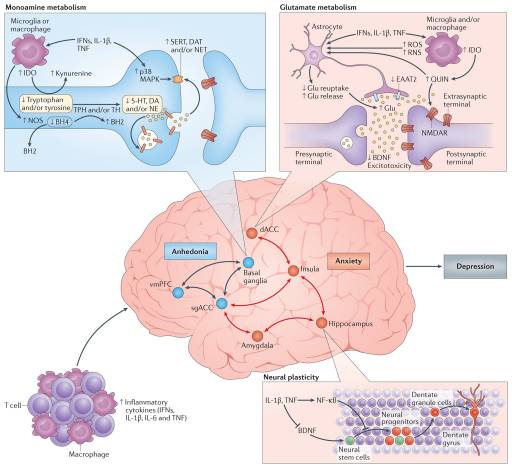
Figure 3: Cytokine targets in the brain: neurotransmitters and neurocircuits. Once in the brain, the inflammatory response can affect metabolic and molecular pathways influencing neurotransmitter systems that can ultimately affect neurocircuits that regulate behaviour, especially behaviours relevant to decreased motivation (anhedonia), avoidance and alarm (anxiety), which characterize several neuropsychiatric disorders including depression. On a molecular level, pro-inflammatory cytokines including type I and II interferons (IFNs), interleukin-1β (IL-1β) and tumour necrosis factor (TNF) can reduce the availability of monoamines — serotonin (5-HT), dopamine (DA) and noradrenaline (NE) — by increasing the expression and function of the presynaptic reuptake pumps (transporters) for 5-HT, DA and NE through activation of mitogen-activated protein kinase (MAPK) pathways and by reducing monoamine synthesis through decreasing enzymatic co-factors such as tetrahydrobiopterin (BH4), which is highly sensitive to cytokine-induced oxidative stress and is involved in the production of nitric oxide (NO) by NO synthase (NOS). Many cytokines, including IFNγ, IL-1β and TNF, can also decrease relevant monoamine precursors by activating the enzyme indoleamine 2,3-dioxygenase (IDO), which breaks down tryptophan, the primary precursor for serotonin, into kynurenine. Activated microglia can convert kynurenine into quinolinic acid (QUIN), which binds to the N-methyl-D-aspartate receptor (NMDAR), a glutamate (Glu) receptor, and together with cytokine-induced reduction in astrocytic Glu reuptake and stimulation of astrocyte Glu release, in part by induction of reactive oxygen species (ROS) and reactive nitrogen species (RNS), can lead to excessive Glu, an excitatory amino acid neurotransmitter. Excessive Glu, especially when binding to extrasynaptic NMDARs, can in turn lead to decreased brain-derived neurotrophic factor (BDNF) and excitotoxicity. Inflammation effects on growth factors such as BDNF in the dentate gyrus of the hippocampus can also affect fundamental aspects of neuronal integrity including neurogenesis, long-term potentiation and dendritic sprouting, ultimately affecting learning and memory. Cytokine effects on neurotransmitter systems, especially DA, can inhibit several aspects of reward motivation and anhedonia in corticostriatal circuits involving the basal ganglia, ventromedial prefrontal cortex (vmPFC) and subgenual and dorsal anterior cingulate cortex (sgACC and dACC, respectively), while also activating circuits regulating anxiety, arousal, alarm and fear including the amygdala, hippocampus, dACC and insula. BH2, dihydrobiopterin; DAT, dopamine transporter; EAAT2, excitatory amino acid transporter 2; NET, noradrenaline transporter; NF-κB, nuclear factor-κB; SERT, serotonin transporter; TH, tyrosine hydroxylase; TPH, tryptophan hydroxylase. Copyrighted 2015. Advanstar. 120580:1115BN.
Effects of Inflammation on Neurocircuitry
Dopamine has a fundamental role in motivation and motor activity, and cytokines have been shown to decrease the release of dopamine in the basal ganglia in association with decreased effort-based motivation as well as reduced activation of reward circuitry in the basal ganglia, in particular the ventral striatum89–91. Inflammatory stimuli have been associated with reductions in reward responsiveness in the striatum across many neuroimaging platforms, demonstrating the validity and reproducibility of these cytokine-mediated effects on the brain in otherwise non-depressed individuals peripherally administered IFNα, endotoxin or typhoid vaccination and imaged by PET, functional magnetic resonance imaging (fMRI), MRS and quantitative magnetization transfer imaging83,89,90,92,93. Interestingly, recent fMRI studies suggest that inflammation-induced decreases in responsiveness to positive reward are also associated with increased sensitivity to aversive stimuli (that is, negative reinforcement) and reduced responsiveness to novelty in the substantia nigra (which is another dopamine-rich structure in the basal ganglia)93,94. Typhoid vaccination has also been shown to activate the subgenual ACC (sgACC), a brain region implicated in depression, and to decrease connectivity of the sgACC with the ventral striatum, an effect modulated by plasma IL-6 (REF. 26). These fMRI findings have recently been extended to patients with depression whose increased plasma CRP level is associated with decreased functional connectivity within reward-related circuits including the ventral striatum and the ventromedial prefrontal cortex that, in turn, mediates the relationship between CRP and anhedonia95. Indeed, patients with depression with a CRP >3 mg L−1 had little, if any, connectivity within reward-related circuits as measured by fMRI, whereas connectivity in patients with depression with a CRP <1 mg L−1 was similar to healthy controls95. Taken together, these data support the notion that the effect of cytokines on the brain in general and dopaminergic pathways in particular lead to a state of decreased motivation or anhedonia, which is a core symptom of depression.
fMRI studies have demonstrated that increased inflammation is also associated with increased activation of threat- and anxiety-related neurocircuitry, including the dACC as well as the insula and amygdala26,96,97. Of note, the dACC and amygdala are regions that exhibit increased activity in patients with high-trait anxiety and neuroticism98, conditions that often accompany depression and are associated with increased inflammation. For example, increased concentrations of oral IL-6 and soluble TNF receptor 2 (also known as TNFRSF1B) in response to a public speaking stressor was significantly correlated with the response of the dACC to a social rejection task97. In addition, increased oral IL-6 expression in response to a social evaluation stressor was significantly correlated with activation of the amygdala, with subjects who exhibited the highest IL-6 responses to stress demonstrating the greatest connectivity within threat circuitry, including the amygdala and the dorsomedial prefrontal cortex, as measured by fMRI99. Interestingly, these data are consistent with the trafficking of monocytes to the amygdala during social defeat stress in mice68.
Risk and Resilience
Increased Inflammation and the Risk for Depression
Both genetic and epigenetic mechanisms may explain why childhood or adult traumas can contribute to exaggerated or persistent inflammation and, ultimately, depression. For example, polymorphisms in CRP were associated not only with increased peripheral blood concentrations of CRP but also with symptoms of post-traumatic stress disorder, especially heightened arousal, in individuals exposed to civilian trauma32. Moreover, gene–environment interactions have been found to influence depression severity in response to chronic interpersonal stress: individuals carrying polymorphisms in IL1B that are associated with higher expression of peripheral IL-1β exhibited more severe depressive symptoms in the context of interpersonal stress than individuals without the IL1B risk allele105. Similarly, mice in which peripheral blood leukocytes produced high concentrations of LPS-induced IL-6 ex vivo before stress exposure showed decreased social exploration after social defeat stress, whereas mice that produced low levels of IL-6 before stress exposure exhibited no behavioural effects in response to social defeat88. Of note, adoptive transfer of bone marrow progenitor cells from mice producing high levels of IL-6 ex vivo to mice that produced low levels of IL-6 made these formerly stress-resilient animals sensitive to the depressive effects of social defeat88.
Epigenetic changes in genes related to inflammation may also affect the risk for depression and anxiety in the context of psychosocial stress. Indeed, the well-documented association of childhood trauma with increased inflammation is linked to stress-induced epigenetic changes in FKBP5, a gene implicated in the development of depression and anxiety as well as in the sensitivity to glucocorticoids106. Allele-specific, childhood trauma-dependent DNA demethylation in functional glucocorticoid response elements of FKBP5 were found to be associated with decreased sensitivity of peripheral blood immune cells to the inhibitory effects of the synthetic glucocorticoid dexamethasone on LPS-induced production of IL-6 in vitro106. Of note, decreased activation of glucocorticoid receptor-responsive genes in association with increased activation of genes regulated by NF-κB has been found to be a ‘fingerprint’ of the effects of chronic stress in several studies examining a variety of psychosocial stressors39,107.
T Cells and Resilience to Depression
Relevant to depression, however, peripheral T cell trafficking in response to glucocorticoids has been shown to be impaired in patients with depression, possibly owing to glucocorticoid resistance as a result of genetically mediated (for example, FKBP5) or inflammasome-mediated mechanisms targeting the glucocorticoid receptor46,115. In addition, inflammatory cytokines and their signalling pathways, including p38 MAPK, have direct inhibitory effects on glucocorticoid receptor function116. Moreover, patients with depression have been shown to have increased numbers of peripheral blood myeloid-derived suppressor cells, which inhibit T cell function117. Of note, activation of the NLRP3 inflammasome leads to increased accumulation of myeloid-derived suppressor cells118. Decreased numbers of peripheral blood TReg cells and reduced concentrations of anti-inflammatory cytokines in the blood, including TGFβ and IL-10, have also been reported in depression119. Thus, it appears that patients with depression may have impairments in neuroprotective and anti-inflammatory T cell responses.
These findings suggest that therapies that boost such T cell responses could be used in patients with depression. Examples include immunization strategies (with CNS antigens as discussed above) that attract T cells to the brain or administration of bacteria, such as Mycobacterium vaccae, or parasites that stimulate TReg cell responses or T cell production of IL-4 (REFS 14,109,110,120). Indeed, colonization of pregnant dams with helminths attenuated the increase of hippocampal IL-1β in neonatal rats infected with bacteria and protected these animals from the subsequent development of microglial sensitization and cognitive dysfunction in adulthood. This effect was associated with increased ex vivo production of IL-4 and decreased production of IL-1β and TNF by splenic macrophages in response to LPS stimulation120. Finally, vagus nerve stimulation could be used to induce anti-inflammatory acetylcholine-producing T cells121. Although many strategies exist to activate anti-inflammatory T cell responses including the induction of TReg cells by administration of mesenchymal stem cells122, the majority of the approaches discussed above have proof-of-concept data in animal models of depression. Nevertheless, the clinical relevance of these approaches has yet to be determined by randomized clinical trials in patients with depression.
Translational Considerations
Guidelines for Anti-Inflammatory Clinical Trials in Depression
- Inflammation only occurs in subgroups of patients with depression30. Clinical trials should enrich for patient populations with evidence of increased inflammation, particularly those identified by a C-reactive protein (CRP) >3 mg L−1, which has been shown to characterize patients with depression with altered reward circuitry and increased basal ganglia glutamate, as well as those who have shown a response to anti-cytokine therapy30,84,95.
- Anti-inflammatory drugs may harm patients without increased inflammation. Inflammatory cytokines and the innate immune response have pivotal roles in synaptic plasticity, neurogenesis, long-term potentiation (which is a fundamental process in learning and memory) and possibly antidepressant response123,128.
- Primary behavioural outcome variables should include measures of anhedonia and anxiety. Neuroimaging studies coupled with studies administering a variety of inflammatory stimuli, including the inflammatory cytokine interferon-α, endotoxin and typhoid vaccination, have revealed that inflammation targets neurocircuits in the brain that regulate motivation and reward as well as anxiety, arousal and alarm35. In addition, these symptoms have been shown to respond to anti-cytokine therapy in limited studies.
- Drugs that specifically target inflammatory cytokines and/or their signalling pathways are preferable. The majority of clinical trials to date have used anti-inflammatory drugs (non-steroidal anti-inflammatory agents and minocycline, a tetracycline antibiotic) that have several off-target effects making the extant data relevant to testing the cytokine hypothesis of depression difficult to interpret31.
- Target engagement must be established in the periphery and ultimately the brain. Protein and gene expression markers of inflammation in the peripheral blood can serve as relevant proxies for inflammation in the brain129, especially given evidence of trafficking of activated peripheral immune cells to the brain in stress-induced animal models of depression. Relevant therapeutic interventions should decrease peripheral inflammatory markers in concert with improvement of specific depressive symptoms. Translocator protein neuroimaging ligands may ultimately serve as direct measures of neuroinflammation and its inhibition by anti-inflammatory therapies in future clinical trials61.

Dr. Alex Jimenez's Insight
When you catch a cold, certain symptoms are triggered by inflammatory markers released in response to illness. While sneezing, coughing and a sore throat serve as the most "obvious" signs you may be sick, what truly keeps you in bed when you have a cold is the accompanying fatigue, inattentiveness, loss of appetite, change in sleep pattern, heightened perception of pain and apathetic withdrawal. These symptoms are similar to the wide array of symptoms that define depression. Many research studies have demonstrated that depression may occur due to an inflammatory response to illness, just as in the case of a common cold. The connection between inflammation and depression has long been argued among healthcare professionals and researchers, where new evidence could open the doors to additional treatment approaches which could help better manage this debilitation health issue.
Conclusion
For glossary and footnotes visit: Ncbi.nlm.nih.gov/pmc/articles/PMC5542678/
Understanding the Phytocannabinoids
Phytocannabinoids in Hemp
Cannabichromene (CBC)
Cannabigerol (CBG)
Phytocannabinoids in Other Crops
Beta-Caryophyllene (BCP)
Diindolylmethane (DIM)
Alkylamides
Falcarinol
Yangonin
Understanding of the endocannabinoid system is growing quickly. As this knowledge does enlarge, science will continue to find more phytocannabinoids in plants and foods that are useful in supporting health in many ways.
In conclusion, many research studies have found a connection between inflammatory pathways and neurocircuits in the brain which can lead to a variety of behavioral responses, such as avoidance and alarm, however, growing evidence has demonstrated that chronic inflammation can lead to depression. Depression is a debilitating disorder which amounts to one of the leading causes of disability worldwide. The article above describes the connection between inflammation and depression. New insights on the results of the research studies could open the possibilities for new treatments to treat depression, among other associated health issues. Moreover, understanding the role of phytocannabinoids in the human body can function as another treatment approach for inflammation associated with depression. Information referenced from the National Center for Biotechnology Information (NCBI). The scope of our information is limited to chiropractic as well as to spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Referenced from: Ncbi.nlm.nih.gov/pmc/articles/PMC5542678/

Additional Topics: Back Pain

