




Pain is a very important function of the human body, including the involvement of nociceptors and the central nervous system, or CNS, to transmit messages from noxious stimulation to the brain. Nociceptors are adrenal glands which are responsible for detecting hazardous or harmful stimuli and transmitting electrical signals into the nervous system. The receptors are present in skin, viscera, muscles, joints and meninges to discover a range of stimulation, which might be mechanical, thermal or chemical.
There are two types of nociceptors:
- C-fibres would be the most common type and are slow to conduct and respond to stimuli. As the proteins in the membrane of the receptor convert the stimulation into electrical impulses that can be taken through the nervous system.
- A-delta fibers are known to conduct more rapidly and convey messages of sharp, momentary pain.
Additionally, there are silent nociceptors which are usually restricted to stimuli but can be "awoken" with high-intensity mechanical stimulation in response to chemical mediators from the body. Nociceptors may have many different voltage-gated stations for transduction that cause a set of action potentials to commence the electric signaling to the nervous system. The excitability and behavior of the cell are based on the types of channels within the nociceptor.
It is important to differentiate between nociception and pain when considering the mechanism of the pain. Nociception is the normal response of the body to noxious stimuli, including reflexes below the suprathreshold that protect the human body from injury. Pain is just perceived when superthreshold for those nociceptors to reach an action possible and initiate the pain pathway is attained, which is comparatively high. The purpose of the article below is to demonstrate the cellular and molecular mechanisms of pain, including acute pain and chronic pain, or persistent pain, as referred to below.
Cellular and Molecular Mechanisms of Pain
Abstract
The nervous system detects and interprets a wide range of thermal and mechanical stimuli as well as environmental and endogenous chemical irritants. When intense, these stimuli generate acute pain, and in the setting of persistent injury, both peripheral and central nervous system components of the pain transmission pathway exhibit tremendous plasticity, enhancing pain signals and producing hypersensitivity. When plasticity facilitates protective reflexes, it can be beneficial, but when the changes persist, a chronic pain condition may result. Genetic, electrophysiological, and pharmacological studies are elucidating the molecular mechanisms that underlie detection, coding, and modulation of noxious stimuli that generate pain.
Introduction: Acute Versus Persistent Pain
The ability to detect noxious stimuli is essential to an organism's survival and wellbeing. This is dramatically illustrated by examination of individuals who suffer from congenital abnormalities that render them incapable of detecting painful stimuli. These people cannot feel piercing pain from a sharp object, heat of an open flame, or even discomfort associated with internal injuries, such as a broken bone. As a result, they do not engage appropriate protective behaviors against these conditions, many of which can be life threatening.
More commonly, alterations of the pain pathway lead to hypersensitivity, such that pain outlives its usefulness as an acute warning system and instead becomes chronic and debilitating. This may be seen, at some level, as an extension of the normal healing process, whereby tissue or nerve damage elicits hyperactivity to promote guarding of the injured area. For example, sunburn produces temporary sensitization of the affected area. As a result normally innocuous stimuli, such as light touch or warmth, are perceived as painful (a phenomenon referred to as allodynia), or normally painful stimuli elicit pain of greater intensity (referred to as hyperalgesia). At its extreme, the sensitization does not resolve. Indeed, individuals who suffer from arthritis, post-herpetic neuralgia (following a bout of shingles), or bone cancer, experience intense and often unremitting pain that is not only physiologically and psychologically debilitating, but may also hamper recovery. Chronic pain may even persist long after an acute injury, perhaps most commonly experienced as lower back pain or sciatica.
Persistent or chronic pain syndromes can be initiated or maintained at peripheral and/or central loci. In either case, the elucidation of molecules and cell types that underlie normal (acute) pain sensation is key to understanding the mechanisms underlying pain hypersensitivity. In the present review we highlight the molecular complexity of the primary afferent nerve fibers that detect noxious stimuli. We not only summarize the processing of acute pain, but also describe how changes in pain processing occur in the setting of tissue or nerve injury.
The profound differences between acute and chronic pain emphasize the fact that pain is not generated by an immutable, hard-wired system, but rather results from the engagement of highly plastic molecules and circuits, the molecular biochemical and neuroanatomical basis of which are the focus of current studies. Importantly, this new information has identified a host of potential therapeutic targets for the treatment of pain. We focus here on the peripheral and second order neurons in the spinal cord; the reader is referred to some excellent reviews of supraspinal pain processing mechanisms, which include remarkable insights that imaging studies have brought to the field (Apkarian et al., 2005).
Anatomical Overview
Nociception is the process by which intense thermal, mechanical or chemical stimuli are detected by a subpopulation of peripheral nerve fibers, called nociceptors (Basbaum and Jessell, 2000). The cell bodies of nociceptors are located in the dorsal root ganglia (DRG) for the body and the trigeminal ganglion for the face, and have both a peripheral and central axonal branch that innervates their target organ and the spinal cord, respectively. Nociceptors are excited only when stimulus intensities reach the noxious range, suggesting that they possess biophysical and molecular properties that enable them to selectively detect and respond to potentially injurious stimuli. There are two major classes of nociceptors. The first includes medium diameter myelinated (Aδ) afferents that mediate acute, well-localized “first” or fast pain. These myelinated afferents differ considerably from the larger diameter and rapidly conducting Aβ fibers that respond to innocuous mechanical stimulation (i.e. light touch). The second class of nociceptor includes small diameter unmyelinated “C” fibers that convey poorly localized, “second” or slow pain.
Electrophysiological studies have further subdivided Aδ nociceptors into two main classes. Type I (HTM: high threshold mechanical nociceptors) respond to both mechanical and chemical stimuli, but have relatively high heat thresholds (>50C). If, however, the heat stimulus is maintained, these afferents will respond at lower temperatures. And most importantly, they will sensitize (i.e. the heat or mechanical threshold will drop) in the setting of tissue injury. Type II Aδ nociceptors have a much lower heat threshold, but a very high mechanical threshold. Activity of this afferent almost certainly mediates the “first” acute pain response to noxious heat. Indeed, compression block of myelinated peripheral nerve fibers eliminates first, but not second, pain. By contrast, the Type I fiber likely mediates the first pain provoked by pinprick and other intense mechanical stimuli.
The unmyelinated C fibers are also heterogeneous. Like the myelinated afferents, most C fibers are polymodal, that is, they include a population that is both heat and mechanically sensitive (CMHs) (Perl, 2007). Of particular interest are the heat responsive, but mechanically insensitive unmyelinated afferents (so-called silent nociceptors) that develop mechanical sensitivity only in the setting of injury (Schmidt et al., 1995). These afferents are more responsive to chemical stimuli (capsaicin or histamine) compared to the CMHs, and likely come into play when the chemical milieu of inflammation alters their properties. Subsets of these afferents are also responsive to a variety of itch-producing pruritogens. It is worth noting that not all C fibers are nociceptors. Some respond to cooling, and a particularly interesting population of unmyelinated afferents responds to innocuous stroking of the hairy skin, but not to heat or chemical stimulation. These latter fibers appear to mediate pleasant touch (Olausson et al., 2008).
Neuroanatomical and molecular characterization of nociceptors has further demonstrated their heterogeneity, particularly for the C fibers (Snider and McMahon, 1998). For example, the so-called ‘peptidergic’ population of C nociceptors releases the neuropeptides, substance P, and calcitonin-gene related peptide (CGRP); they also express the TrkA neurotrophin receptor, which responds to nerve growth factor (NGF). The non-peptidergic population of C nociceptors expresses the c-Ret neurotrophin receptor that is targeted by glial-derived neurotrophic factor (GDNF), as well as neurturin and artemin. A large percentage of the c-Ret-positive population also binds the IB4 isolectin, and expresses G protein-coupled receptors of the Mrg family (Dong et al., 2001), as well as specific purinergic receptor subtypes, notably P2X3. Nociceptors can also be distinguished according to their differential expression of channels that confer sensitivity to heat (TRPV1), cold (TRPM8), acidic milieu (ASICs), and a host of chemical irritants (TRPA1) (Julius and Basbaum, 2001). As noted below, these functionally and molecularly heterogeneous classes of nociceptors associate with specific function in the detection of distinct pain modalities.
The Nociceptor: a Bidirectional Signaling Machine
One generally thinks of the nociceptor as carrying information in one direction, transmitting noxious stimuli from the periphery to the spinal cord. However, primary afferent fibers have a unique morphology, called pseudo-unipolar, wherein both central and peripheral terminals emanate from a common axonal stalk. The majority of proteins synthesized by the DRG or trigeminal ganglion cell are distributed to both central and peripheral terminals. This distinguishes the primary afferent neuron from the prototypical neuron, where the recipient branch of the neuron (the dendrite) is biochemically distinct from the transmission branch (the axon). The biochemical equivalency of central and peripheral terminals means that the nociceptor can send and receive messages from either end. For example, just as the central terminal is the locus of Ca2+-dependent neurotransmitter release, so the peripheral terminal releases a variety of molecules that influence the local tissue environment. Neurogenic inflammation, in fact, refers to the process whereby peripheral release of the neuropeptides, CGRP and substance P, induces vasodilation and extravasation of plasma proteins, respectively (Basbaum and Jessell, 2000). Furthermore, whereas only the peripheral terminal of the nociceptor will respond to environmental stimuli (painful heat, cold and mechanical stimulation), both the peripheral and central terminals can be targeted by a host of endogenous molecules (such as pH, lipids, and neurotransmitters) that regulate its sensitivity. It follows that therapeutics directed at both terminals can be developed to influence the transmission of pain messages. For example, spinal (intrathecal) delivery of morphine targets opioid receptors expressed by the central terminal of nociceptors, whereas topically applied drugs (such as local anesthetics or capsaicin) regulate pain via an action at the peripheral terminal.
Central Projections of the Nociceptor
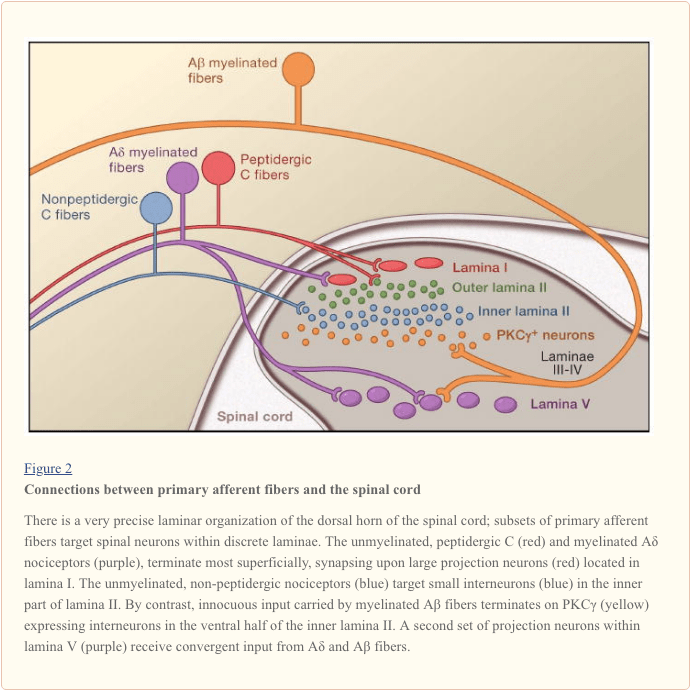
Primary afferent nerve fibers project to the dorsal horn of the spinal cord, which is organized into anatomically and electrophysiological distinct laminae (Basbaum and Jessell, 2000) (Figure 1). For example, Aδ nociceptors project to lamina I as well as to deeper dorsal horn (lamina V). The low threshold, rapidly conducting Aβ afferents, which respond to light touch, project to deep laminae (III, IV, and V). By contrast, C nociceptors project more superficially to laminae I and II.
This remarkable stratification of afferent subtypes within the superficial dorsal horn is further highlighted by the distinct projection patterns of C nociceptors (Snider and McMahon, 1998). For example, most peptidergic C fibers terminate within lamina I and the most dorsal part of lamina II. By contrast, the nonpeptidergic afferents, including the Mrg-expressing subset, terminate in the mid-region of lamina II. The most ventral part of lamina II is characterized by the presence of excitatory interneurons that express the gamma isoform of protein kinase C (PKC), which has been implicated in injury-induced persistent pain (Malmberg et al., 1997). Recent studies indicate that this PKCγ layer is targeted predominantly by myelinated non-nociceptive afferents (Neumann et al., 2008). Consistent with these anatomical studies, electrophysiological analyses demonstrate that spinal cord neurons within lamina I are generally responsive to noxious stimulation (via Aδ and C fibers), neurons in laminae III and IV are primarily responsive to innocuous stimulation (via Aβ), and neurons in lamina V receive a convergent non-noxious and noxious input via direct (monosynaptic) Aδ and Aβ inputs and indirect (polysynaptic) C fiber inputs. The latter are called wide dynamic range (WDR) neurons, in that they respond to a broad range of stimulus intensities. There is also commonly a visceral input to these WDR neurons, such that the resultant convergence of somatic and visceral likely contributes to the phenomenon of referred pain, whereby pain secondary to an injury affecting a visceral tissue (for example, the heart in angina) is referred to a somatic structure (for example, the shoulder).
Ascending Pathways and the Supraspinal Processing of Pain
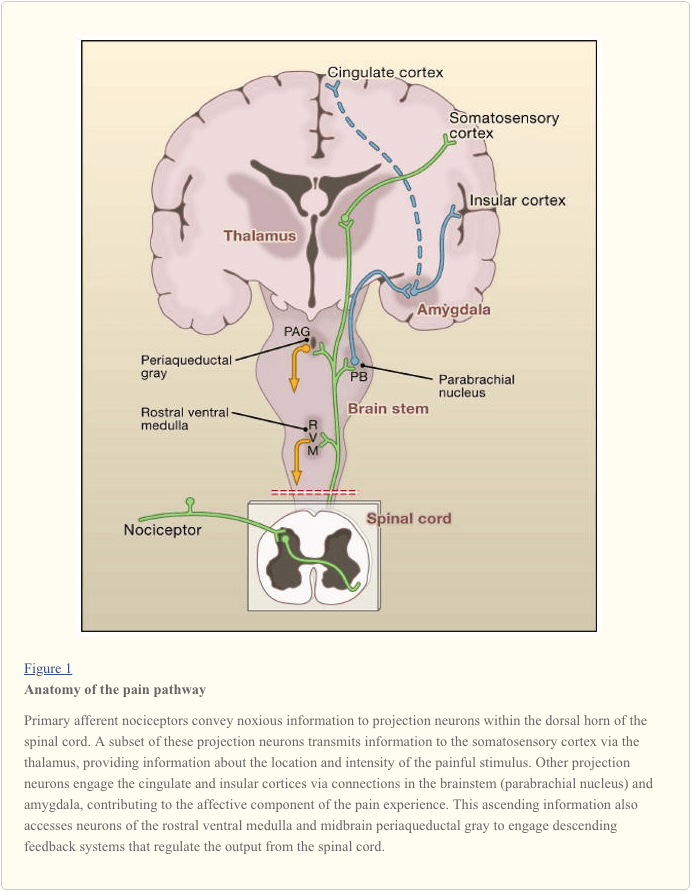
Projection neurons within laminae I and V constitute the major output from the dorsal horn to the brain (Basbaum and Jessell, 2000). These neurons are at the origin of multiple ascending pathways, including the spinothalamic and spinoreticulothalamic tracts, which carry pain messages to the thalamus and brainstem, respectively (Figure 2). The former is particularly relevant to the sensory-discriminative aspects of the pain experience (that is, where is the stimulus and how intense is it?), whereas the latter may be more relevant to poorly localized pains. More recently, attention has focused on spinal cord projections to the parabrachial region of the dorsolateral pons, because the output of this region provides for a very rapid connection with the amygdala, a region generally considered to process information relevant to the aversive properties of the pain experience.
From these brainstem and thalamic loci, information reaches cortical structures. There is no single brain area essential for pain (Apkarian et al., 2005). Rather, pain results from activation of a distributed group of structures, some of which are more associated with the sensory-discriminative properties (such as the somatosensory cortex) and others with the emotional aspects (such as the anterior cingulate gyrus and insular cortex). More recently, imaging studies demonstrate activation of prefrontal cortical areas, as well as regions not generally associated with pain processing (such as the basal ganglia and cerebellum). Whether and to what extent activation of these regions is more related to the response of the individual to the stimulus, or to the perception of the pain is not clear. Finally, Figure 2 illustrates the powerful descending controls that influence (both positive and negatively) the transmission of pain messages at the level of the spinal cord.
Acute Pain
The primary afferent nerve fiber detects environmental stimuli (of a thermal, mechanical, or chemical nature) and transduces this information into the language of the nervous system, namely electrical current. First, we review progress in understanding the molecular basis of signal detection, and follow this with a brief overview of recent genetic studies that highlight the contribution of voltage-gated channels to pain transmission (Figure 3).
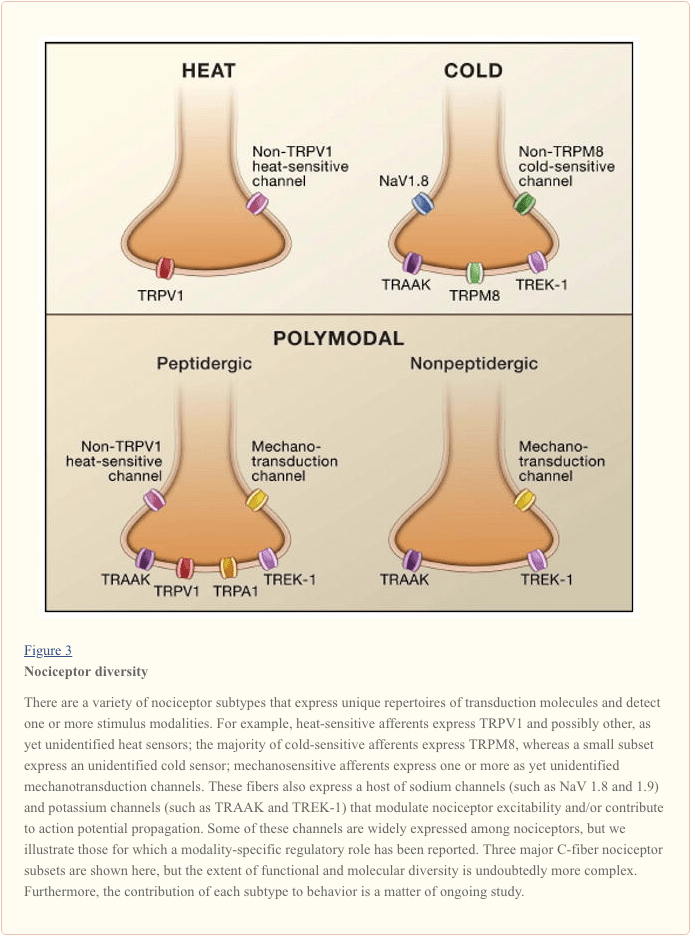
Activating the Nociceptor: Heat
Human psychophysical studies have shown that there is a clear and reproducible demarcation between the perception of innocuous warmth and noxious heat, which enables us to recognize and avoid temperatures capable of causing tissue damage. This pain threshold, which typically rests around 43°C, parallels the heat sensitivity of C and Type II Aδ nociceptors described earlier. Indeed, cultured neurons from dissociated dorsal root ganglia show similar heat sensitivity. The majority display a threshold of 43°C, with a smaller cohort activated by more intense heat (threshold >50°C) (Cesare and McNaughton, 1996; Kirschstein et al., 1997; Leffler et al., 2007; Nagy and Rang, 1999). Molecular insights into the process of heat sensation came from the cloning and functional characterization of the receptor for capsaicin, the main pungent ingredient in ‘hot’ chili peppers. Capsaicin and related vanilloid compounds produce burning pain by depolarizing specific subsets of C and Aδ nociceptors through activation of the capsaicin (or vanilloid) receptor, TRPV1, one of approximately 30 members of the greater transient receptor potential (TRP) ion channel family (Caterina et al., 1997). The cloned TRPV1 channel is also gated by increases in ambient temperature, with a thermal activation threshold (∼43°C).
Several lines of evidence support the hypothesis that TRPV1 an endogenous transducer of noxious heat. First, TRPV1 is expressed in the majority of heat-sensitive nociceptors (Caterina et al., 1997). Second, capsaicin- and heat-evoked currents are similar, if not identical, in regard to their pharmacological and biophysical properties, as are those of heterologously expressed TRPV1 channels. Third, and as described in greater detail below, TRPV1-evoked responses are markedly enhanced by pro-algesic or pro-inflammatory agents (such as extracellular protons, neurotrophins, or bradykinin), all of which produce hypersensitivity to heat in vivo (Tominaga et al., 1998)). Fourth, analysis of mice lacking this ion channel not only revealed a complete loss of capsaicin sensitivity, but these animals also exhibit significant impairment in their ability to detect and respond to noxious heat (Caterina et al., 2000; Davis et al., 2000). These studies also demonstrated an essential role for this channel in the process whereby tissue injury and inflammation leads to heat hypersensitivity, reflecting the ability of TRPV1 to serve as a molecular integrator of thermal and chemical stimuli (Caterina et al., 2000; Davis et al., 2000).
The contribution of TRPV1 to acute heat sensation, however, has been challenged by data collected from an ex vivo preparation in which recordings are obtained from the soma of DRG neurons with intact central and peripheral fibers. In one study, no differences were observed in heat-evoked responses from wild type and TRPV1-deficient animals (Woodbury et al., 2004), but a more recent analysis from this group found that TRPV1-deficient mice do, indeed, lack a cohort of neurons robustly activated by noxious heat (Lawson et al., 2008). Taken together with the results described above we conclude that TRPV1 unquestionably contributes to acute heat sensation, but agree that TRPV1 is not solely responsible for heat transduction.
In this regard, whereas TRPV1-deficient mice lack a component of behavioral heat sensitivity, the use of high dose capsaicin to ablate the central terminals of TRPV1-expressing primary afferent fibers results in a more profound, if not complete loss of acute heat pain sensitivity (Cavanaugh et al., 2009). As for the TRPV1 mutant, there is also a loss of tissue injury-evoked heat hyperalgesia. Taken together these results indicate that both the TRPV1-dependent and TRPV1-independent component of noxious heat sensitivity is mediated via TRPV1-expressing nociceptors.
What accounts for the TRPV1-independent component of heat sensation? A number of other TRPV channel subtypes, including TRPV2, 3 and 4, have emerged as candidate heat transducers that could potentially cover detection of stimulus intensities flanking that of TRPV1, including both very hot (>50°C) and warm (mid-30°Cs) temperatures (Lumpkin and Caterina, 2007). Heterologously expressed TRPV2 channels display a temperature activation threshold of ∼52°C, whereas TRPV3 and TRPV4 are activated between 25 - 35°C. TRPV2 is expressed in a subpopulation of Aδ neurons that respond to high threshold noxious heat and its biophysical properties resemble those of native high threshold heat-evoked currents (Leffler et al., 2007; Rau et al., 2007). As yet, there are no published reports describing either physiological or behavioral tests of TRPV2 knockout mice. On the other hand, TRPV3- and TRPV4-deficient mice do display altered thermal preference when placed on a surface of graded temperatures, suggesting that these channels contribute in some way to temperature detection in vivo (Guler et al., 2002). Interestingly, both TRPV3 and TRPV4 show substantially greater expression in keratinocytes and epithelial cells compared to sensory neurons, raising the possibility that detection of innocuous heat stimuli involves a functional interplay between skin and the underlying primary afferent fibers (Chung et al., 2003; Peier et al., 2002b).
Activating the Nociceptor: Cold
As for capsaicin and TRPV1, natural cooling agents, such as menthol and eucalyptol, have been exploited as pharmacological probes to identify and characterize cold-sensitive fibers and cells (Hensel and Zotterman, 1951; Reid and Flonta, 2001) and the molecules that underlie their behavior. Indeed, most cold-sensitive neurons respond to menthol and display a thermal activation threshold of ∼25°C. TRPM8 is a cold and menthol-sen sitive channel whose physiological characteristics match those of native cold currents and TRPM8-deficient mice show a very substantial loss of menthol and cold-evoked responses at the cellular or nerve fiber level. Likewise, these animals display severe deficits in cold-evoked behavioral responses (Bautista et al., 2007; Colburn et al., 2007; Dhaka et al., 2007) over a wide range of temperatures spanning 30 to 10°C. As in the case of TRPV1 and he at, TRPM8-deficient mice are not completely insensitive to cold. For example, there remains a small (∼4%) cohort of cold-sensitive, menthol-insensitive neurons that have a low threshold of activation, of approximately 12°C. These may account for the residual cold sensitivity seen in behavioral tests, wherein TRPM8-deficient animals can still avoid extremely cold surfaces below 10°C. Importantly, TRPM8-deficient mice show normal sensitivity to noxious heat. Indeed, TRPV1 and TRPM8 are expressed in largely non-overlapping neuronal populations, consistent with the notion that hot and cold detection mechanisms are organized into anatomically and functionally distinct ‘labeled lines.’
Based on heterologous expression systems, TRPA1 has also been suggested to detect cold, specifically within the noxious (<15°C) range. Moreover TRPA1 is activated by the cooling compounds icilin and menthol (Bandell et al., 2004; Karashima et al., 2007; Story et al., 2003), albeit at relatively high concentrations compared to their actions at TRPM8. However, there continues to be disagreement as to whether native or recombinant TRPA1 are intrinsically cold sensitive (Bandell et al., 2004; Jordt et al., 2004; Karashima et al., 2009; Nagata et al., 2005; Zurborg et al., 2007). This controversy has not been resolved by the analysis of two independent TRPA1-deficient mouse lines. At the cellular level, one study showed normal cold-evoked responses in TRPA1-deficient neurons following a 30 second drop in temperature from 22°C to 4 °C (Bautista et al., 2006); a more recent study has shown a decrease in cold sensitive neurons from 26% (WT) to 10% (TRPA1-/-), when tested after a 200 sec drop in temperature, from 30°C to 10°C (Karashima et al., 2009). In behavioral studies, TRPA1-deficient mice display responses similar to wild-type littermates in the cold-plate and acetone-evoked evaporative cooling assays (Bautista et al., 2006). A second study using the same assays showed that female, but not male, TRPA1 knockout animals displayed attenuated cold sensitivity compared to wild type littermates (Kwan et al., 2006). Karashima et al found no difference in shivering or paw withdrawal latencies in male or female TRPA1-deficient mice on the cold plate test, but observed that prolonged exposure to the cold surface elicited jumping in wild type, but not TRPA1-deficient animals (Karashima et al., 2009). Conceivably, the latter phenotype reflects a contribution of TRPA1 to cold sensitivity in the setting of tissue injury, but not to acute cold pain. Consistent with the latter hypothesis, single nerve fiber recordings show no decrement in acute cold sensitivity in TRPA1-deficient mice (Cavanaugh et al., 2009; Kwan et al., 2009). Finally, it is noteworthy that capsaicin-treated mice lacking the central terminals of TRPV1-expressing fibers show intact behavioral responses to cool and noxious cold stimuli (Cavanaugh et al., 2009). Because TRPA1 is expressed in a subset of TRPV1-positive neurons, it follows that TRPA1 is not required for normal acute cold sensitivity. Future studies using mice deficient for both TRPM8 and TRPA1 will help to resolve these issues and to identify the molecules and cell types that underlie the residual TRPM8-independent component of cold sensitivity.
Additional molecules, including voltage-gated sodium channels (discussed below), voltage-gated potassium channels, and two-pore background KCNK potassium channels, coordinate with TRPM8 to fine tune cold thresholds or to propagate cold-evoked action potentials (Viana et al., 2002; Zimmermann et al., 2007; Noel et al., 2009). For example, specific Kv1 inhibitors increase the temperature threshold of cold-sensitive neurons and injection of these inhibitors into the rodent hindpaw reduces behavioral responses to cold, but not to heat or mechanical stimuli (Madrid et al., 2009). Two members of the KCNK channel family, KCNK2 (TREK-1) and KCNK4 (TRAAK) are expressed in a subset of C-fiber nociceptors (Noel et al., 2009) and can be modulated by numerous physiological and pharmacological stimuli, including pressure and temperature. Furthermore, mice lacking these channels display abnormalities in sensitivity to pressure, heat, and cold (Noel et al., 2009). Although these findings suggest that TREK-1 and TRAAK channels modulate nociceptor excitability, it remains unclear how their intrinsic sensitivity to physical stimuli relates to their in vivo contribution to thermal or mechanical transduction.
Activating the Nociceptor: Mechanical
The somatosensory system detects quantitatively and qualitatively diverse mechanical stimuli, ranging from light brush of the skin to distension of the bladder wall. A variety of mechanosensitive neuronal subtypes are specialized to detect this diverse array of mechanical stimuli and can be categorized according to threshold sensitivity. High threshold mechanoreceptors include C fibers and slowly adapting Aδ mechanoreceptor (AM) fibers, both of which terminate as free nerve endings in the skin. Low threshold mechanoreceptors include Aδ D-hair fibers that terminate on down hairs in the skin and detect light touch. Finally, Aβ fibers that innervate Merkel cells, Pacinian corpuscles and hair follicles detect texture, vibration, and light pressure.
As in the case of thermal stimuli, mechanical sensitivity has been probed at a number of levels, including dissociated sensory neurons in culture, ex-vivo fiber recordings, as well as recordings from central (i.e. dorsal horn neurons) and measurements of behavioral output. Ex-vivo skin-nerve recordings have been most informative in matching stimulus properties (such as intensity, frequency, speed, and adaptation) to specific fiber subtypes. For example, Aβ fibers are primarily associated with sensitivity to light touch, whereas C and Aδ fibers are primarily responsive to noxious mechanical insults. At the behavioral level, mechanical sensitivity is typically assessed using two techniques. The most common involves measuring reflex responses to constant force applied to the rodent hind paw by calibrated filaments (Von Frey hairs). The second applies increasing pressure to the paw or tail via a clamp system. In either case, information about mechanical thresholds is obtained under normal (acute) or injury (hypersensitivity) situations. One of the challenges in this area has been to develop additional behavioral assays that measure different aspects of mechanosensation, such as texture discrimination and vibration, which will facilitate the study of both noxious and non-noxious touch (Wetzel et al., 2007).
At the cellular level, pressure can be applied to the cell bodies of cultured somatosensory neurons (or to their neurites) using a glass probe, changes in osmotic strength, or stretch via distension of an elastic culture surface, though it is unclear which stimulus best mimics physiological pressure (Bhattacharya et al., 2008; Cho et al., 2006; Cho et al., 2002; Drew et al., 2002; Hu and Lewin, 2006; Lin et al., 2009; Takahashi and Gotoh, 2000). Responses can be assessed using electrophysiological or live cell imaging methods. The consensus from such studies is that that pressure opens a mechanosensitive cation channel to elicit rapid depolarization. However, a dearth of specific pharmacological probes and molecular markers with which to characterize these responses or to label relevant neuronal subtypes has hampered attempts to match cellular activities with anatomically or functionally defined nerve fiber subclasses. These limitations have also impeded the molecular analysis of mechansosensation and the identification of molecules that constitute the mechanotransduction machinery. Nonetheless, a number of candidates have emerged, based largely on studies of mechanosensation in model genetic organisms. Mammalian orthologues of these proteins have been examined using gene targeting approaches in mice, in which the techniques mentioned above can be used to assess deficits in mechanosensation at all levels. Below we briefly summarize some of the candidates revealed in these studies.
Candidate Mechanotransducers: DEG/ENaC Channels
Studies in the nematode Caenorhabditis elegans (C. elegans) have identified mec-4 and mec-10, members of the degenerin/epithelial Na+ channel (DEG/ENaC) families, as mechanotransducers in body touch neurons (Chalfie, 2009). Based on these studies, the mammalian orthologues ASIC 1, 2 and 3 have been proposed as mechanotransduction channels. ASICs are acid-sensitive ion channels that serve as receptors for extracellular protons (tissue acidosis) produced during ischemia (see below). Although these channels are expressed by both low and high threshold mechanosensitive neurons, genetic studies do not uniformly support an essential role in mechanotransduction. Mice lacking functional ASIC1 channels display normal behavioral responses to cutaneous touch, and little or no change in mechanical sensitivity when assessed by single fiber recording (Page et al., 2004; Price et al., 2000). Likewise, peripheral nerve fibers from ASIC2-deficient mice display only a slight decrease in action potential firing to mechanical stimuli, whereas ASIC3-deficient fibers display a slight increase (no change in mechanical thresholds or baseline behavioral mechanical sensitivity was observed in these animals) (Price et al., 2001; Roza et al., 2004). Analysis of mice deficient for both ASIC2 and ASIC3 also fails to support a role for these channels in cutaneous mechanotransduction (Drew et al., 2004). Thus, although these channels appear to play a role in musculoskeletal and ischemic pain (see below), their contribution to mechanosensation remains unresolved.
Genetic studies suggest that C. elegans mec-4/mec-10 channels exist in a complex with the stomatin-like protein MEC-2 (Chalfie, 2009). Mice lacking the MEC-2 orthologue, SLP3, display a loss of mechanosensitivity in low-threshold Aβ and Aδ fibers, but not in C fibers (Wetzel et al., 2007). These mice exhibit altered tactile acuity, but display normal responses to noxious pressure, suggesting that SLP3 contributes to the detection of innocuous, but not noxious mechanical stimuli. Whether SLP3 functions in a mechanotransduction complex or interacts with ASICs in mammalian sensory neurons is unknown.
Candidate Mechanotransducers: TRP Channels
As noted above, when expressed heterologously, TRPV2 not only responds to noxious heat, but also to osmotic stretch. Additionally, native TRPV2 channels in vascular smooth muscle cells are activated by direct suction and osmotic stimuli (Muraki et al., 2003). A role for TRPV2 for somatosensory mechanotransduction in vivo has not yet been tested.
TRPV2 is robustly expressed in medium and large diameter, Aδ fibers that respond to both mechanical and thermal stimuli (Caterina et al., 1999; Muraki et al., 2003). TRPV4 shows modest expression in sensory ganglia, but is more abundantly expressed in the kidney and stretch-sensitive urothelial cells of the bladder (Gevaert et al., 2007; Mochizuki et al., 2009). When heterologously expressed, both TRPV2 and TRPV4 have been shown to respond to changes in osmotic pressure (Guler et al., 2002; Liedtke et al., 2000; Mochizuki et al., 2009; Strotmann et al., 2000). Analysis of TRPV4-deficient animals suggests a role in osmosensation as knockout animals display defects in blood pressure, water balance, and bladder voiding (Gevaert et al., 2007; Liedtke and Friedman, 2003). These animals exhibit normal acute cutaneous mechanosensation, but show deficits in models of mechanical and thermal hyperalgesia (Alessandri-Haber et al., 2006; Chen et al., 2007; Grant et al., 2007; Suzuki et al., 2003). Thus, TRPV4 is unlikely to serve as a primary mechanotransducer in sensory neurons, but may contribute to injury-evoked pain hypersensitivity.
TRPA1 has also been proposed to serve as a detector of mechanical stimuli. Heterologously expressed mammalian TRPA1 is activated by membrane crenators (Hill and Schaefer, 2007) and the worm orthologue is sensitive to mechanical pressure applied via a suction pipette (Kindt et al., 2007). However, TRPA1-deficient mice display only weak defects in mechanosensory behavior and the results are inconsistent. Two studies reported no change in mechanical thresholds in TRPA1-deficient animals (Bautista et al., 2006; Petrus et al., 2007), whereas a third study reported deficits (Kwan et al., 2006). A more recent study shows that C and Aβ mechanosensitive fibers in TRPA1 knockout animals have altered responses to mechanical stimulation (some increased and others decreased) (Kwan et al., 2009). Whether and how these differential physiological effects are manifest at the level of behavior is unclear. Taken together, TRPA1 does not appear to function as a primary detector of acute mechanical stimuli, but perhaps modulates excitability of mechanosensitive afferents.
Candidate Mechanotransducers: KCNK Channels
In addition to the potential mechanotransducer role of KCNK2 and 4 (see above), KCNK18 has been discussed for its possible contribution to mechanosensation. Thus, KCNK18 is targeted by hydroxy-a-sanshool, the pungent ingredient in Szechuan peppercorns that produces tingling and numbing sensations, suggestive of an interaction with touch-sensitive neurons (Bautista et al., 2008; Bryant and Mezine, 1999; Sugai et al., 2005). KCNK18 is expressed in a subset of presumptive peptidergic C fibers and low threshold (Aβ) mechanoreceptors, where it serves as a major regulator of action potential duration and excitability (Bautista et al., 2008; Dobler et al., 2007). Moreover, sanshool depolarizes osmo- and mechanosensitive large diameter sensory neurons, as well as a subset of nociceptors (Bautista et al., 2008; Bhattacharya et al., 2008). Although it is not known if KCNK18 is directly sensitive to mechanical stimulation, it may be a critical regulator of the excitability of neurons involved in innocuous or noxious touch sensation.
In summary, the molecular basis of mammalian mechanotransduction is far from clarified. Mechanical hypersensitivity in response to tissue or nerve injury represents a major clinical problem and thus elucidating the biological basis of touch under normal and pathophysiological conditions remains one of the main challenges in somatosensory and pain research.
Activating the Nociceptor: Chemical
Chemo-nociception is the process by which primary afferent neurons detect environmental irritants and endogenous factors produced by physiological stress. In the context of acute pain, chemo-nociceptive mechanisms trigger aversive responses to a variety of environmental irritants. Here, again, TRP channels have prominent roles, which is perhaps not surprising given that they function as receptors for plant-derived irritants, including capsaicin (TRPV1), menthol (TRPM8), as well as the pungent ingredients in mustard and garlic plants, isothiocyanates and thiosulfinates (TRPA1) (Bandell et al., 2004; Caterina et al., 1997; Jordt et al., 2004; McKemy et al., 2002; Peier et al., 2002a).
With respect to environmental irritants, TRPA1 has emerged as a particularly interesting member of this group. This is because TRPA1 responds to compounds that are structurally diverse but unified in their ability to form covalent adducts with thiol groups. For example, allyl isothiocyanate (from wasabi) or allicin (from garlic) are membrane permeable electrophiles that activate TRPA1 by covalently modifying cysteine residues within the amino-terminal cytoplasmic domain of the channel (Hinman et al., 2006; Macpherson et al., 2007). How this promotes channel gating is currently unknown. Nevertheless, simply establishing the importance of thiol reactivity in this process has implicated TRPA1 as a key physiological target for a wide and chemically diverse group of environmental toxicants. One notable example is acrolein (2-propenal), a highly reactive α,β-unsaturated aldehyde present in tear gas, vehicle exhaust, or smoke from burning vegetation (i.e. forest fires and cigarettes). Acrolein and other volatile irritants (such as hypochlorite, hydrogen peroxide, formalin, and isocyanates) activate sensory neurons that innervate the eyes and airways, producing pain and inflammation (Bautista et al., 2006; Bessac and Jordt, 2008; Caceres et al., 2009). This action can have especially dire consequences for those suffering from asthma, chronic cough, or other pulmonary disorders. Mice lacking TRPA1 show greatly reduced sensitivity to such agents, underscoring the critical nature of this channel as a sensory detector of reactive environmental irritants (Caceres et al., 2009). In addition to these environmental toxins, TRPA1 is targeted by some general anesthetics (such as isofluorane) or metabolic byproducts of chemotherapeutic agents (such as cyclophosphamide), which likely underlies some of the adverse side effects of these drugs, including acute pain and robust neuroinflammation (Bautista et al., 2006; Matta et al., 2008).
Finally, chemical irritants and other pro-algesic agents are also produced endogenously in response to tissue damage or physiological stress, including oxidative stress. Such factors can act alone, or in combination, to sensitize nociceptors to thermal and/or mechanical stimuli, thereby lowering pain thresholds. The result of this action is to enhance guarding and protective reflexes in the aftermath of injury. Thus, chemo-nociception represents an important interface between acute and persistent pain, especially in the context of peripheral tissue injury and inflammation, as discussed in greater detail below.
Acute Pain: Conducting the Pain Signal
Once thermal and mechanical signals are transduced by the primary afferent terminal, the receptor potential activates a variety of voltage-gated ion channels. Voltage-gated sodium and potassium channels are critical to the generation of action potentials that convey nociceptor signals to synapses in the dorsal horn. Voltage-gated calcium channels play a key role in neurotransmitter release from central or peripheral nociceptor terminals to generate pain or neurogenic inflammation, respectively. We restrict our discussion to members of the sodium and calcium channel families that serve as targets of currently used analgesic drugs, or for which human genetics support a role in pain transmission. A recent review has discussed the important contribution of KCNQ potassium channels, including the therapeutic benefit of increasing K+ channel activity for the treatment of persistent pain (Brown and Passmore, 2009).
Voltage-Gated Sodium Channels
A variety of sodium channels are expressed in somatosensory neurons, including the tetrodotoxin (TTX)-sensitive channels Nav1.1, 1.6 and 1.7, and the TTX-insensitive channels, Nav1.8 and 1.9. In recent years, the contribution of Nav1.7 has received much attention, as altered activity of this channel leads to a variety of human pain disorders (Cox et al., 2006; Dib-Hajj et al., 2008). Patients with loss-of-function mutations within this gene are unable to detect noxious stimuli, and as a result suffer injuries due to lack of protective reflexes. In contrast, a number of gain-of-function mutations in Nav1.7 leads to hyperexcitability of the channel and are associated with two distinct pain disorders in humans, erythromelalgia, and paroxysmal extreme pain disorder, both of which cause intense burning sensations (Estacion et al., 2008; Fertleman et al., 2006; Yang et al., 2004). Animal studies have demonstrated that Nav1.7 is highly upregulated in a variety of inflammatory pain models. Indeed, analysis of mice lacking Nav1.7 in C nociceptors supports a key role for this channel in mechanical and thermal hypersensitivity following inflammation, and in acute responses to noxious mechanical stimuli (Nassar et al., 2004). Somewhat surprisingly, pain induced by nerve injury is unaltered, suggesting that distinct sodium channel subtypes, or another population of Nav1.7-expressing afferents, contribute to neuropathic pain (Nassar et al., 2005).
The Nav1.8 sodium channel is also highly expressed by most C nociceptors. As with Nav1.7 knockout animals, those lacking Nav1.8 display modest deficits in sensitivity to innocuous or noxious heat, or innocuous pressure; however, they display attenuated responses to noxious mechanical stimuli (Akopian et al., 1999). Nav1.8 is also required for the transmission of cold stimuli, as mice lacking this channel are insensitive to cold over a wide range of temperatures (Zimmermann et al., 2007). This is because Nav1.8 is unique among voltage-sensitive sodium channels in that it does not inactivate at low temperature, making it the predominant action potential generator under cold conditions.
Interestingly, transgenic mice lacking the Nav1.8 expressing subset of sensory neurons, which were deleted by targeted expression of diphtheria toxin A (Abrahamsen et al., 2008), display attenuated responses to both low and high threshold mechanical stimuli and cold. In addition, mechanical and thermal hypersensitivity in inflammatory pain models is severely attenuated. The differential phenotypes of mice lacking Nav1.8 channels versus deletion of the Nav1.8-expressing neurons presumably reflects the contribution of multiple voltage-gated sodium channel subtypes to transmission of pain messages.
Voltage-gated sodium channels are targets of local anesthetic drugs, highlighting the potential for the development of subtype-specific analgesics. Nav1.7 is a particularly interesting target for treating inflammatory pain syndromes, in part, because the human genetic studies suggest that Nav1.7 inhibitors should reduce pain without altering other essential physiological processes (see above). Another potential application of sodium channel blockers may be to treat extreme hypersensitivity to cold, a particularly troublesome adverse side effect of platinum-based chemotherapeutics, such as oxaliplatin (Attal et al., 2009). Nav1.8 (or TRPM8) antagonists may alleviate this, or other forms of cold allodynia. Finally, the great utility of the antidepressant serotonin and norepinephrine reuptake inhibitors for the treatment of neuropathic pain may, in fact, result from their ability to block voltage gated sodium channels (Dick et al., 2007).
Voltage-Gated Calcium Channels
A variety of voltage-gated calcium channels are expressed in nociceptors. N-, P/Q- and T-type calcium channels have received the most attention. P/Q-type channels are expressed at synaptic terminals in laminae II-IV of the dorsal horn. Their exact role in nociception is not completely resolved. However, mutations in these channels have been linked to familial hemiplegic migraine (de Vries et al., 2009). N- and T-type calcium channels are also expressed by C-fibers and are upregulated under pathophysiological states, as in models of diabetic neuropathy or after other forms of nerve injury. Animals lacking Cav2.2 or 3.2 show reduced sensitization to mechanical or thermal stimuli following inflammation or nerve injury, respectively (Cao, 2006; Swayne and Bourinet, 2008; Zamponi et al., 2009; Messinger et al., 2009). Moreover, ω-conotoxin GVIA, which blocks N-type channels, is administered intrathecally (as ziconotide) to provide relief for intractable cancer pain (Rauck et al., 2009).
All calcium channels are heteromeric proteins composed of α1 pore forming subunits and the modulatory subunits α2δ, α2β or α2γ. The α2δ subunit regulates current density and kinetics of activation and inactivation. In C nociceptors, the α2δ subunit is dramatically upregulated following nerve injury and plays a key role in injury-evoked hypersensitivity and allodynia (Luo et al., 2001). Indeed, this subunit is the target of gabapentinoid class of anticonvulsants, which are now widely used to treat neuropathic pain (Davies et al., 2007).
Persistent Pain: Peripheral Mechanisms
Persistent pain associated with injury or diseases (such as diabetes, arthritis, or tumor growth) can result from alterations in the properties of peripheral nerves. This can occur as a consequence of damage to nerve fibers, leading to increased spontaneous firing or alterations in their conduction or neurotransmitter properties. In fact, the utility of topical and even systemic local anesthetics for the treatment of different neuropathic pain conditions (such as postherpetic neuralgia) likely reflects their action on sodium channels that accumulate in injured nerve fibers.
The Chemical Milieu of Inflammation
Peripheral sensitization more commonly results from inflammation-associated changes in the chemical environment of the nerve fiber (McMahon et al., 2008). Thus, tissue damage is often accompanied by the accumulation of endogenous factors released from activated nociceptors or non-neural cells that reside within or infiltrate into the injured area (including mast cells, basophils, platelets, macrophages, neutrophils, endothelial cells, keratinocytes, and fibroblasts). Collectively. these factors, referred to as the ‘inflammatory soup’, represent a wide array of signaling molecules, including neurotransmitters, peptides (substance P, CGRP, bradykinin), eicosinoids and related lipids (prostaglandins, thromboxanes, leukotrienes, endocannabinoids), neurotrophins, cytokines, and chemokines, as well as extracellular proteases and protons. Remarkably, nociceptors express one or more cell surface receptors capable of recognizing and responding to each of these pro-inflammatory or pro-algesic agents (Figure 4). Such interactions enhance excitability of the nerve fiber, thereby heightening its sensitivity to temperature or touch.
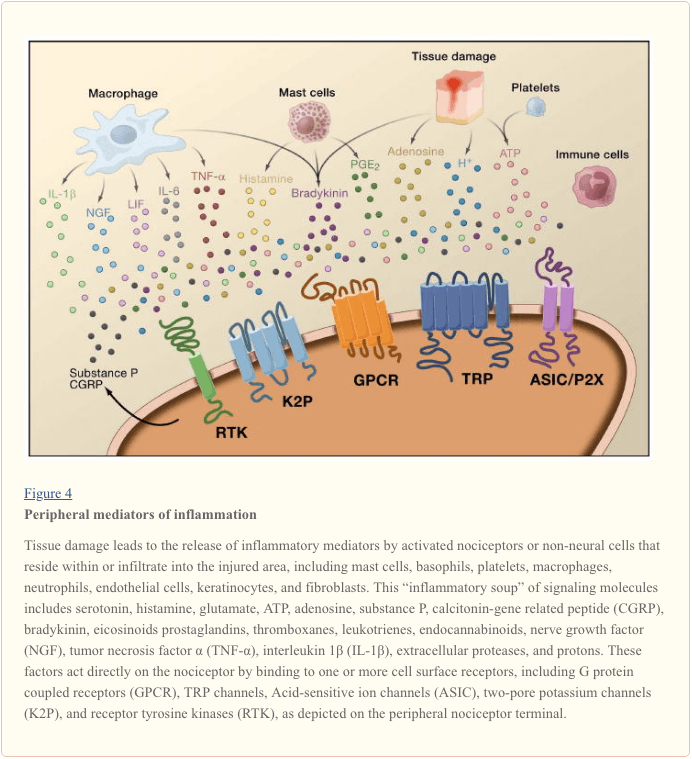
Unquestionably the most common approach to reducing inflammatory pain involves inhibiting the synthesis or accumulation of components of the inflammatory soup. This is best exemplified by non-steroidal anti-inflammatory drugs, such as aspirin or ibuprofen, which reduce inflammatory pain and hyperalgesia by inhibiting cyclooxygenases (Cox-1 and Cox-2) involved in prostaglandin synthesis. A second approach is to block the actions of inflammatory agents at the nociceptor. Here, we highlight examples that provide new insight into cellular mechanisms of peripheral sensitization, or which form the basis of new therapeutic strategies for treating inflammatory pain.
NGF is perhaps best known for its role as a neurotrophic factor required for survival and development of sensory neurons during embryogenesis, but in the adult, NGF is also produced in the setting of tissue injury and constitutes an important component of the inflammatory soup (Ritner et al., 2009). Among its many cellular targets, NGF acts directly on peptidergic C fiber nociceptors, which express the high affinity NGF receptor tyrosine kinase, TrkA, as well as the low affinity neurotrophin receptor, p75 (Chao, 2003; Snider and McMahon, 1998). NGF produces profound hypersensitivity to heat and mechanical stimuli through two temporally distinct mechanisms. At first, a NGF-TrkA interaction activates downstream signaling pathways, including phospholipase C (PLC), mitogen-activated protein kinase (MAPK), and phosphoinositide 3-kinase (PI3K). This results in functional potentiation of target proteins at the peripheral nociceptor terminal, most notably TRPV1, leading to a rapid change in cellular and behavioral heat sensitivity (Chuang et al., 2001). In addition to these rapid actions, NGF is also retrogradely transported to the nucleus of the nociceptor, where it promotes increased expression of pro-nociceptive proteins, including substance P, TRPV1, and the Nav1.8 voltage-gated sodium channel subunit (Chao, 2003; Ji et al., 2002). Together, these changes in gene expression enhance excitability of the nociceptor and amplify the neurogenic inflammatory response.
In addition to neurotrophins, injury promotes the release of numerous cytokines, chief among them interleukin 1β (IL-1β) and IL-6, and tumor necrosis factor α (TNF-α) (Ritner et al., 2009). Although there is evidence to support a direct action of these cytokines on nociceptors, their primary contribution to pain hypersensitivity results from potentiation of the inflammatory response and increased production of pro-algesic agents (such as prostaglandins, NGF, bradykinin, and extracellular protons).
Irrespective of their pro-nociceptive mechanisms, interfering with neurotrophin or cytokine signaling has become a major strategy for controlling inflammatory disease or resulting pain. The main approach involves blocking NGF or TNF-α action with a neutralizing antibody. In the case of TNF-α, this has been remarkably effective in the treatment of numerous autoimmune diseases, including rheumatoid arthritis, leading to dramatic reduction in both tissue destruction and accompanying hyperalgesia (Atzeni et al., 2005). Because the main actions of NGF on the adult nociceptor occur in the setting of inflammation, the advantage of this approach is that hyperalgesia will decrease without affecting normal pain perception. Indeed, anti-NGF antibodies are currently in clinical trials for treatment of inflammatory pain syndromes (Hefti et al., 2006).
Targets of the Inflammatory Soup
TRPV1. Robust hypersensitivity to heat can develop with inflammation or after injection of specific components of the inflammatory soup (such as bradykinin or NGF). Lack of such sensitization in TRPV1-deficient mice provides genetic support for the idea that TRPV1 is a key component of the mechanism through which inflammation produces thermal hyperalgesia (Caterina et al., 2000; Davis et al., 2000). Indeed, in vitro studies have shown that TRPV1 functions as a polymodal signal integrator whose thermal sensitivity can be profoundly modulated by components of the inflammatory soup (Tominaga et al., 1998). Some of these inflammatory agents (for example, extracellular protons and lipids) function as direct positive allosteric modulators of the channel, whereas others (bradykinin, ATP, and NGF) bind to their own receptors on primary afferents and modulate TRPV1 through activation of downstream intracellular signaling pathways. In either case, these interactions result in a profound decrease in the channel's thermal activation threshold, as well as an increase in the magnitude of responses at supra-threshold temperatures—the biophysical equivalents of allodynia and hyperalgesia, respectively.
However, there remains controversy concerning the intracellular signaling mechanisms most responsible for TRPV1 modulation (Lumpkin and Caterina, 2007). Reminiscent of ancestral TRP channels in the fly eye, many mammalian TRP channels are activated or positively modulated by phospholipase C-mediated cleavage of plasma membrane phosphatidyl inositol 4,5 bisphosphate (PIP2). Of course, there are many downstream consequences of this action, including a decrease in membrane PIP2, increase levels of diacylglycerol and its metabolites, increased cytoplasmic calcium, as well as consequent activation of protein kinases. In the case of TRPV1, most, if not all, of these pathways have been implicated in the sensitization process and it remains to be seen which are most relevant to behavioral thermal hypersensitivity. Nevertheless, there is broad agreement that TRPV1 modulation is relevant to tissue injury-evoked pain hypersensitivity, particularly in the setting of inflammation. This would include conditions such as sunburn, infection, rheumatoid or osteoarthritis, and inflammatory bowl disease. Another interesting example includes pain from bone cancer (Honore et al., 2009), where tumor growth and bone destruction are accompanied by extremely robust tissue acidosis, as well as production of cytokines, neurotrophins, and prostaglandins.
TRPA1. As described above, TRPA1 is activated by compounds that form covalent adducts with cysteine residues. In addition to environmental toxins, this includes endogenous thiol reactive electrophiles that are produced during tissue injury and inflammation, or as a consequence of oxidative or nitrative stress. Chief among such agents are 4-hydroxy-2-nonenal and 15-deoxy-Δ12,14-prostaglandin J2, which are both α,β unsaturated aldehydes generated through peroxidation or spontaneous dehydration of lipid second messengers (Andersson et al., 2008; Cruz-Orengo et al., 2008; Materazzi et al., 2008; Trevisani et al., 2007). Other endogenous TRPA1 agonists include nitrooleic acid, hydrogen peroxide, and hydrogen sulfide. In addition to these directly acting agents, TRPA1 is also modulated indirectly by pro-algesic agents, such as bradykinin, which act via PLC-coupled receptors. Indeed, TRPA1-deficient mice show dramatically reduced cellular and behavioral responses to all of these agents, as well as a reduction in tissue injury-evoked thermal and mechanical hypersensitivity (Bautista et al., 2006; Kwan et al., 2006). Finally, because TRPA1 plays a key role in neurogenic and other inflammatory responses to both endogenous agents and volatile environmental toxins, its contribution to airway inflammation, such as occurs in asthma, is of particular interest. Indeed, genetic or pharmacological blockade of TRPA1 reduces airway inflammation in a rodent model of allergen-evoked asthma (Caceres et al., 2009).
ASICs. As noted above, ASIC channels are members of the DEG/ENaC family that are activated by acidification, and thus represent another important site for the action of extracellular protons produced as a consequence of tissue injury or metabolic stress. ASIC subtypes can form a variety of homomeric or heteromeric channels, each having distinct pH sensitivity and expression profile. Channels containing the ASIC3 subtype are specifically expressed by nociceptors and especially well represented in fibers that innervate skeletal and cardiac muscle. In these tissues, anaerobic metabolism leads to buildup of lactic acid and protons, which activate nociceptors to generate musculoskeletal or cardiac pain (Immke and McCleskey, 2001). Interestingly, ASIC3-containing channels open in response to the modest decrease in pH (e.g. 7.4 to 7.0) that occurs with cardiac ischemia (Yagi et al., 2006). Lactic acid also significantly potentiates proton-evoked gating through a mechanism involving calcium chelation (Immke and McCleskey, 2003). Thus, ASIC3-containing channels detect and integrate signals specifically associated with muscle ischemia and, in this way, are functionally distinct from other acid sensors on the primary afferent, such as TRPV1 or other ASIC channel subtypes.
Persistent Pain: Central Mechanisms
Central sensitization refers to the process through which a state of hyperexcitability is established in the central nervous system, leading to enhanced processing of nociceptive (pain) messages (Woolf, 1983). Although numerous mechanisms have been implicated in central sensitization here we focus on three: alteration in glutamatergic neurotransmission/NMDA receptor-mediated hypersensitivity, loss of tonic inhibitory controls (disinhibition) and glial-neuronal interactions (Figure 5).
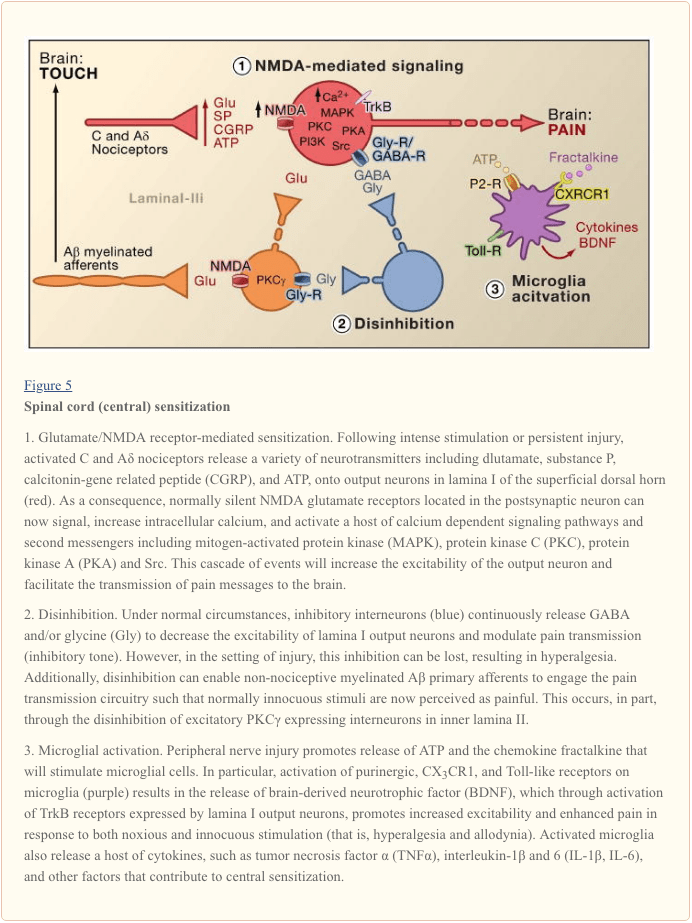
Glutamate/NMDA Receptor-Mediated Sensitization
Acute pain is signaled by the release of glutamate from the central terminals of nociceptors, generating excitatory post-synaptic currents (EPSCs) in second order dorsal horn neurons. This occurs primarily through activation of postsynaptic AMPA and kainate subtypes of ionotropic glutamate receptors. Summation of sub-threshold EPSCs in the postsynaptic neuron will eventually result in action potential firing and transmission of the pain message to higher order neurons. Under these conditions, the NMDA subtype of glutamate channel is silent, but in the setting of injury, increased release of neurotransmitters from nociceptors will sufficiently depolarize postsynaptic neurons to activate quiescent NMDA receptors. The consequent increase in calcium influx can strengthen synaptic connections between nociceptors and dorsal horn pain transmission neurons, which in turn will exacerbate responses to noxious stimuli (that is, generate hyperalgesia).
In many ways, this processes is comparable to that implicated in the plastic changes associated with hippocampal long-term potentiation (LTP) (for a review on LTP in the pain pathway, see Drdla and Sandkuhler, 2008). Indeed, drugs that block spinal LTP reduce tissue injury-induced hyperalgesia. As in the case of hippocampal LTP, spinal cord central sensitization is dependent on NMDA-mediated elevations of cytosolic Ca2+ in the postsynaptic neuron. Concurrent activation of metabotropic glutamate and substance P receptors on the postsynaptic neuron may also contribute to sensitization by augmenting cytosolic calcium. Downstream activation of a host of signaling pathways and second messenger systems, notably kinases (such as MAPK, PKA, PKC, PI3K, Src), further increases excitability of these neurons, in part by modulating NMDA receptor function (Latremoliere and Woolf, 2009). Illustrative of this model is the demonstration that spinal injections of a nine amino acid peptide fragment of Src not only disrupts an NMDA receptor–Src interaction but also markedly decreases the hypersensitivity produced by peripheral injury, without changing acute pain. Src null mutant mice also display reduced mechanical allodynia after nerve injury (Liu et al., 2008).
In addition to enhancing inputs from the site of injury (primary hyperalgesia), central sensitization contributes to the condition in which innocuous stimulation of areas surrounding the injury site can produce pain. This secondary hyperalgesia involves heterosynaptic facilitation, wherein inputs from Aβ afferents, which normally respond to light touch, now engage pain transmission circuits, resulting in profound mechanical allodynia. The fact that compression block of peripheral nerve fibers concurrently interrupts conduction in Aβ afferents and eliminates secondary hyperalgesia indicates that these abnormal circuits are established in clinical settings as well as in animal models (Campbell et al., 1988).
Loss of GABAergic and Glycinergic Controls: Disinhibition
GABAergic or glycinergic inhibitory interneurons are densely distributed in the superficial dorsal horn and are at the basis of the longstanding gate control theory of pain, which postulates that loss of function of these inhibitory interneurons (disinhibition) would result in increased pain (Melzack and Wall, 1965). Indeed, in rodents, spinal administration of GABA (bicuculline) or glycine (strychnine) receptor antagonists (Malan et al., 2002; Sivilotti and Woolf, 1994; Yaksh, 1989) produces behavioral hypersensitivity resembling that observed after peripheral injury. Consistent with these observations, peripheral injury leads to a decrease in inhibitory postsynaptic currents in superficial dorsal horn neurons. Although Moore et al. (2002) suggested that the disinhibition results from peripheral nerve injury-induced death of GABAergic interneurons, this claim has been contested (Polgar et al., 2005). Regardless of the etiology, the resulting decreased tonic inhibition enhances depolarization and excitation of projection neurons. As for NMDA-mediated central sensitization, disinhibition enhances spinal cord output in response to painful and non-painful stimulation, contributing to mechanical allodynia (Keller et al., 2007; Torsney and MacDermott, 2006).
Following upon an earlier report that deletion of the gene encoding PKCγ in the mouse leads to a marked decrease in nerve injury-evoked mechanical hypersensitivity (Malmberg et al., 1997), recent studies address the involvement of these neurons in the disinhibitory process. Thus, after blockade of glycinergic inhibition with strychnine, innocuous brushing of the hindpaw activates PKCγ-positive interneurons in lamina II (Miraucourt et al., 2007), as well as projection neurons in lamina I. Because PKCγ-positive neurons in the spinal cord are located only in the innermost part of lamina II (Figure 1), it follows that these neurons are essential for the expression of nerve injury-evoked persistent pain, and that disinhibitory mechanisms lead to their hyperactivation.
Other studies indicate that changes in the projection neuron, itself, contribute to the dis-inhibitory process. For example, peripheral nerve injury profoundly down-regulates the K+-Cl- co-transporter KCC2, which is essential for maintaining normal K+ and Cl- gradients across the plasma membrane (Coull et al., 2003). Downregulating KCC2, which is expressed in lamina I projection neurons, results in a shift in the Cl- gradient, such that activation of GABA-A receptors depolarize, rather than hyperpolarize the lamina I projection neurons. This would, in turn, enhance excitability and increase pain transmission. Indeed, pharmacological blockade or siRNA-mediated downregulation of KCC2 in the rat induces mechanical allodynia. Nonetheless, Zeilhofer and colleagues suggest that, even after injury, sufficient inhibitory tone remains such that enhancement of spinal GABAergic neurotransmission might be a valuable approach to reduce pain hypersensitivity induced by peripheral nerve injury (Knabl et al., 2008). In fact, studies in mice suggest that drugs specifically targeting GABAA complexes containing α2 and/or α3 subunits reduce inflammatory and neuropathic pain without producing sedative-hypnotic side effects typically associated with benzodiazepines, which enhance activity of α1-containing channels.
Disinhibition can also occur through modulation of glycinergic signaling. In this case the mechanism involves a spinal cord action of prostaglandins (Harvey et al., 2004). Specifically, tissue injury induces spinal release of the prostaglandin, PGE2, which acts on EP2 receptors expressed by excitatory interneurons and projection neurons in the superficial dorsal horn. Resultant stimulation of the cAMP-PKA pathway phosphorylates GlyRa3 glycine receptor subunits, rendering the neurons unresponsive to the inhibitory effects of glycine. Accordingly, mice lacking the GlyRa3 gene have decreased heat and mechanical hypersensitivity in models of tissue injury.
Glial-Neuronal Interactions
Finally, glial cells, notably microglia and astrocytes, also contribute to the central sensitization process that occurs in the setting of injury. Under normal conditions, microglia function as resident macrophages of the central nervous system. They are homogeneously distributed within the grey matter of the spinal cord and are presumed to function as sentinels of injury or infection. Within hours of peripheral nerve injury, however, microglia accumulate in the superficial dorsal horn within the termination zone of injured peripheral nerve fibers. Microglia also surround the cell bodies of ventral horn motoneurons, whose peripheral axons are concurrently damaged. The activated microglia release a panoply of signaling molecules, including cytokines (such as TNF-α, interleukin-1β and 6), which enhance neuronal central sensitization and nerve injury-induced persistent pain (DeLeo et al., 2007). Indeed, injection of activated brain microglia into the cerebral spinal fluid at the level of the spinal cord can reproduce the behavioral changes observed after nerve injury (Coull et al., 2005). Thus, it appears that microglial activation is sufficient to trigger the persistent pain condition (Tsuda et al., 2003).
As microglia are activated following nerve, but not inflammatory tissue injury, it follows that activation of the afferent fiber, which occurs under both injury conditions, is not the critical trigger for microglial activation. Rather, physical damage of the peripheral afferent must induce the release of specific signals that are detected by microglia. Chief among these is ATP, which targets microglial P2-type purinergic receptors. Of particular interest are P2X4 (Tsuda et al., 2003), P2X7 (Chessell et al., 2005) and P2Y12 (Haynes et al., 2006; Kobayashi et al., 2008) receptor subtypes. Indeed, ATP was used to activate brain microglia in the spinal cord transplant studies referred to above (Tsuda et al., 2003). Furthermore, genetic or pharmacological blockade of purinergic receptor function (Chessell et al., 2005; Tozaki-Saitoh et al., 2008; Ulmann et al., 2008) prevents or reverses nerve injury-induced mechanical allodynia (Honore et al., 2006; Kobayashi et al., 2008; Tozaki-Saitoh et al., 2008; Tsuda et al., 2003).
Coull and colleagues proposed a model in which ATP/P2X4-mediated activation of microglia triggers a mechanism of disinhibition (Coull et al., 2005). Specifically, they demonstrated that ATP-evoked activation of P2X4 receptors induces release of the brain-derived neurotrophic factor (BDNF) from microglia. The BDNF, in turn, acts upon TrkB receptors on lamina I projection neurons, to generate a change in the Cl- gradient, which as described above, would shift the action of GABA from hyperpolarization to depolarization. Whether the BDNF-induced effect involves KCC2 expression, as occurs after nerve injury, is not known. Regardless of the mechanism, the net result is that activation of microglia will sensitize lamina I neurons such that their response to monosynaptic inputs from nociceptors, or indirect inputs from Aβ afferents, is enhanced.
In addition to BDNF, activated microglia, like peripheral macrophages, release and respond to numerous chemokines and cytokines, and these also contribute to central sensitization. For example, in the uninjured (normal) animal, the chemokine fractalkine (CXCL1) is expressed by both primary afferents and spinal cord neurons (Lindia et al., 2005; Verge et al., 2004; Zhuang et al., 2007). In contrast, the fractalkine receptor (CX3CR1) is expressed on microglial cells and importantly, is upregulated after peripheral nerve injury (Lindia et al., 2005; Zhuang et al., 2007). Because spinal delivery of fractalkine can activate microglia, it appears that nerve injury-induced release of fractalkine provides yet another route through which microglia can be engaged in the process of central sensitization. Indeed blockade of CX3CR1 with a neutralizing antibody prevents both the development and maintenance of injury-induced persistent pain (Milligan et al., 2004; Zhuang et al., 2007). This pathway may also be part of a positive feedback loop through which injured nerve fibers and microglial cells interact in a reciprocal and recurrent fashion to amplify pain signals. This point is underscored by the fact that fractalkine must be cleaved from the neuronal surface prior to signaling, an action that is carried out by the microglial-derived protease, cathepsin S, inhibitors of which reduce nerve injury-induced allodynia and hyperalgesia (Clark et al., 2007). Importantly, spinal administration of cathepsin S generates behavioral hypersensitivity in wild type, but not in CX3CX1 knockout mice, linking cathepsin S to fractalkine signaling (Clark et al., 2007; Zhuang et al., 2007). Although the factor(s) that initiates release of cathepsin S from microglia remains to be determined,. ATP seems a reasonable possibility.
Very recently, several members of the Toll-like receptors (TLRs) family have also been implicated in the activation of microglia following nerve injury. TLRs are transmembrane signaling proteins expressed in peripheral immune cells and glia. As part of the innate immune system, they recognize molecules that are broadly shared by pathogens. Genetic or pharmacological inhibition of TLR2, TLR3 or TLR4 function in mice results not only in decreased microglial activation, but also reduces the hypersensitivity triggered by peripheral nerve injury (Kim et al., 2007; Obata et al., 2008; Tanga et al., 2005). Unknown are the endogenous ligands that activate TLR2-4 after nerve-injury. Among the candidates are mRNAs or heat shock proteins that could leak from the damaged primary afferent neurons and diffuse into the extracellular milieu of the spinal cord.
The contribution of astrocytes to central sensitization is less clear. Astrocytes are unquestionably induced in the spinal cord after injury to either tissue or nerve (for a review, see Ren and Dubner, 2008). But, in contrast to microglia, astrocyte activation is generally delayed and persists much longer, up to several months. One interesting possibility is that astrocytes are more critical to the maintenance, rather than to the induction of central sensitization and persistent pain.
Finally, it is worth noting that peripheral injury not only activates glia in the spinal cord, but also in the brainstem, where glia contribute to supraspinal facilitatory influences on the processing of pain messages in the spinal cord (see Figure 2), a phenomenon named descending facilitation (for a review, see Ren and Dubner, 2008). Such facilitation is especially prominent in the setting of injury, and appears to counteract the feedback inhibitory controls that concurrently arise from various brainstem loci (Porreca et al., 2002).

Dr. Alex Jimenez's Insight
As established by the International Association for the Study of Pain, or the IASP, pain is "an unpleasant sensory and emotional experience associated with acutal or potential tissue damage, or described in terms of tissue damage or both. Numerous research studies have been proposed to demonstrate the physiological basis of pain, however, none has been able to include the entire aspects associated with pain perception. Understanding the pain mechanisms of acute pain versus chronic pain is fundamental during clinical evaluations as this can help determine the best treatment approach for patients with underlying health issues.
Specificity in the Transmission and Control of Pain Messages
Understanding how stimuli are encoded by the nervous system to elicit appropriate behaviors is of fundamental importance to the study of all sensory systems. In the simplest form, a sensory system uses labeled lines to transduce stimuli and elicit behaviors through strictly segregated circuits. This is perhaps best exemplified by the taste system, where exchanging a sweet receptor for a bitter one in a population of “sweet taste afferents” does not alter the behavior provoked by activity in that labeled line; under these conditions, a bitter tastant stimulates these afferents to elicit a perception of sweetness (Mueller et al., 2005).
In the pain pathway, there is also evidence to support the existence of labeled lines. As mentioned above, heat and cold are detected by largely distinct subsets of primary afferent fibers. Moreover, elimination of subsets of nociceptors can produce selective deficits in the behavioral response to a particular noxious modality. For example, destruction of TRPV1-expressing nociceptors produces a profound loss of heat pain (including heat hyperalgesia), with no change in sensitivity to painful mechanical or cold stimuli. Conversely, deletion of the MrgprD subset of nociceptors results in a highly selective deficit in mechanical responsiveness, with no change in heat sensitivity (Cavanaugh et al., 2009). Further evidence for functional segregation at the level of the nociceptor comes from the analysis of two different opioid receptor subtypes (Scherrer et al., 2009). Specifically, the mu opioid receptor (MOR) predominates in the peptidergic population, whereas the delta opioid receptor (DOR) is expressed in non-peptidergic nociceptors. MOR-selective agonists block heat pain, whereas DOR selective agonists block mechanical pain, again illustrating functional separation of molecularly distinct nociceptor populations.
These observations argue for behaviorally-relevant specificity at the level of the nociceptor. However, this is likely to be an oversimplification for at least two reasons. First, many nociceptors are polymodal and can therefore be activated by thermal, mechanical, or chemical stimuli, leaving one to wonder how elimination of large cohorts of nociceptors can have modality-specific effects. This argues for a substantial contribution of spinal circuits to the process whereby nociceptive signals are encoded into distinct pain modalities. Indeed, an important future goal is to better delineate neuronal subtypes within the dorsal horn and characterize their synaptic interactions with functionally or molecularly defined subpopulations of nociceptors. Second, the pain system shows a tremendous capacity for change, particularly in the setting of injury, raising questions about whether and how a labeled line system might accommodate such plasticity, and how alterations in such mechanisms underlie maladaptive changes that produce chronic pain. Indeed, we know that substance P-saporin-mediated deletion of a discrete population of lamina I dorsal horn neurons, which express the substance P receptor, can reduce both the thermal and mechanical pain hypersensitivity that occurs after tissue or nerve injury (Nichols et al., 1999). Such observations suggest that in the setting of injury specificity of the labeled line is not strictly maintained as information is transmitted to higher levels of the neuraxis.
Clearly, answers to these questions will require the combined use of anatomical, electrophysiological, and behavioral methods to map the physical and functional circuitry that underlies nociception and pain. The ongoing identification of molecules and genes that mark specific neuronal cell types (both peripheral and central) provides essential tools with which to manipulate genetically or pharmacologically these neurons and to link their activities to specific components of pain behavior under normal and pathophysiological circumstances. Doing so should bring us closer to understanding how acute pain gives way to the maladaptive changes that produce chronic pain, and how this switch can be prevented or reversed.

Hemp vs Marijuana: What's the Difference?
With approximately half of U.S. states now permitting the sale of medical marijuana, and a few even allowing the sale of marijuana for recreational use, more and more people are becoming interested in the possible health benefits of this controversial plant.
Whilst science on its medical use continues to advance, many people these days are considering how they could access the plant's health benefits without experiencing its well-known unwanted psychoactive effect. This is completely possible with marijuana's close relative, hemp but it is essential that you be aware of the difference so that you may be a smart consumer.
One Cultivars of the Exact Same Plant
Fundamentally, both hemp and marijuana are the exact same plant: Cannabis sativa. There is evidence that Cannabis sativa L has been grown in Asia thousands of years back for its fiber as well as a food supply. Humans eventually realized that the flowering tops of the plant had psychoactive properties. With time, as humans have done so with many other plants, Cannabis farmers began cultivating specific plants to enhance specific properties.
Nowadays, though some might argue the true number of plant types, there are really two simple distinctions,
Hemp - A plant primarily cultivated outside the United States, although a few U.S. countries let it be grown for study purposes) for use in clothes, paper, biofuels, bioplastics, dietary supplements, cosmetics, and foods. Hemp is cultivated outdoors as a large crop with both male and female plants being present to boost pollination and improve seed production. Legally imported industrial hemp contains less than 0.3 percent of its carcinogenic chemical tetrahydrocannabinol, or THC, content. In reality, legally imported hemp will usually specifically eliminate any extracts in the plant's dried flowering tops.
Marijuana (Marihuana) - Cannabis sativa especially cultivated to enhance its THC content to be used for medicinal or recreational purposes. Marijuana plants are typically grown indoors, under controlled conditions, and growers eliminate all of the male plants from the harvest to prevent fertilization because fertilization lowers the plant's THC degree.
Legality of Medical Marijuana
The medical use of marijuana is an increased area of controversy for researchers and consumers alike. Although maybe not quite half of U.S. states have legalized the medical use of this plant, it remains illegal under federal law, and consequently its use remains controversial regardless of the fact that there does seem to be real health benefits for various serious health issues.
Those that are looking to use marijuana for medical use should talk about its benefits versus its dangers with a skilled health-care professional before using it. In addition, many consumers who have an interest in its health benefits do not need the psychoactive side effects of THC or the danger of a positive drug test.
Hemp: Health Benefits without the Risks
Imported hemp, that has a very low, almost absent, level of THC, can be a solution for consumers that are looking for the plant's health benefits minus the effects of THC.
Though THC has some health benefits, hemp comprises more than 80 bioactive compounds that could provide excellent support for a range of health issues, such as stress response, positive mood, and physical discomfort or pain. Hemp can also benefit gastrointestinal health, help keep a healthy inflammatory response throughout the entire body, and support normal immune function.
If you are considering the use of a nutritional supplement product which includes hemp, then it's ideal to buy a product from a trusted source.
In conclusion, both the peripheral and central nervous system detect, interpret and regulate a wide range of thermal and mechanical stimulation as well as environmental and endogenous chemical irritants. If the stimuli is too intense, it can generate acute pain where in the instance of persistent or chronic pain, pain transmission can be tremendously affected. The article above describes the cellular and molecular mechanisms of pain for guidance in clinical assessments. Furthermore, the use of hemp can have many health benefits in comparison to the controversial effects of marijuana. Information referenced from the National Center for Biotechnology Information (NCBI). The scope of our information is limited to chiropractic as well as to spinal injuries and conditions. To discuss the subject matter, please feel free to ask Dr. Jimenez or contact us at 915-850-0900 .
Curated by Dr. Alex Jimenez
Referenced from: Ncbi.nlm.nih.gov

Additional Topics: Back Pain
Back pain is one of the most prevalent causes for disability and missed days at work worldwide. As a matter of fact, back pain has been attributed as the second most common reason for doctor office visits, outnumbered only by upper-respiratory infections. Approximately 80 percent of the population will experience some type of back pain at least once throughout their life. The spine is a complex structure made up of bones, joints, ligaments and muscles, among other soft tissues. Because of this, injuries and/or aggravated conditions, such as herniated discs, can eventually lead to symptoms of back pain. Sports injuries or automobile accident injuries are often the most frequent cause of back pain, however, sometimes the simplest of movements can have painful results. Fortunately, alternative treatment options, such as chiropractic care, can help ease back pain through the use of spinal adjustments and manual manipulations, ultimately improving pain relief.
